

Protein implicated in nonsyndromic mental retardation regulates protein kinase A (PKA) activity
- PMID: 22375002
- PMCID: PMC3340277
- DOI: 10.1074/jbc.M111.261875
Protein implicated in nonsyndromic mental retardation regulates protein kinase A (PKA) activity
Abstract
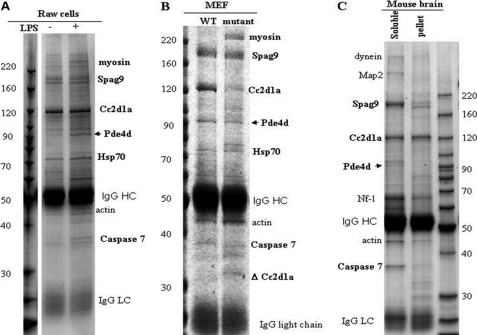
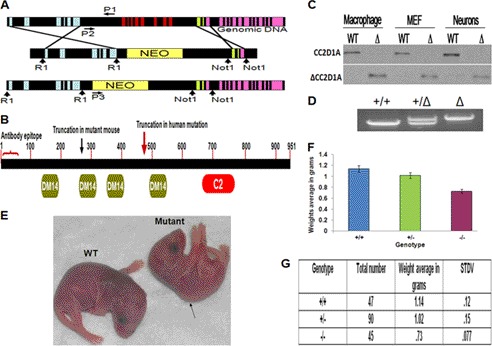
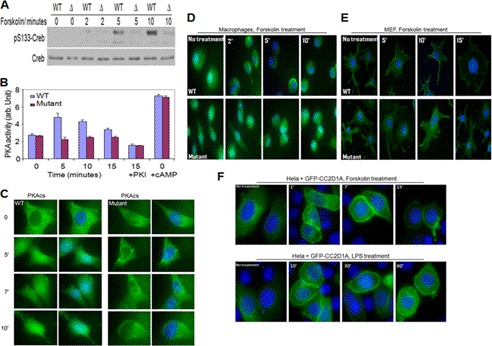
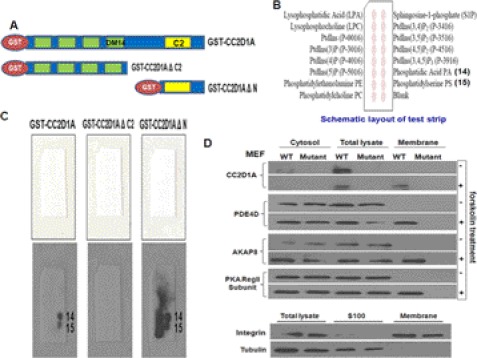
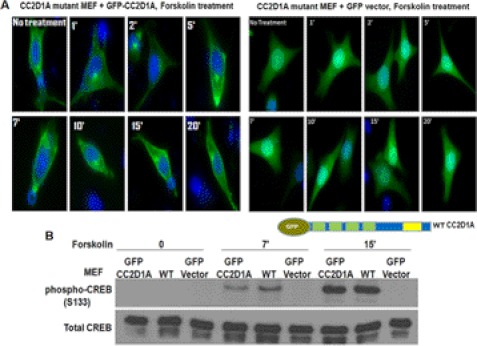
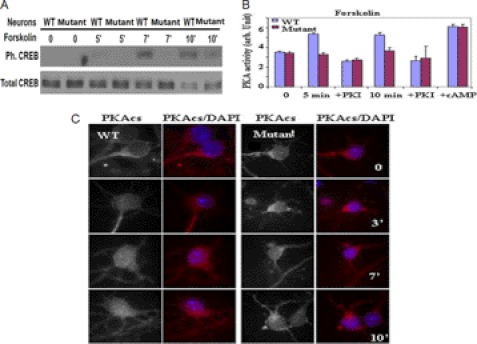
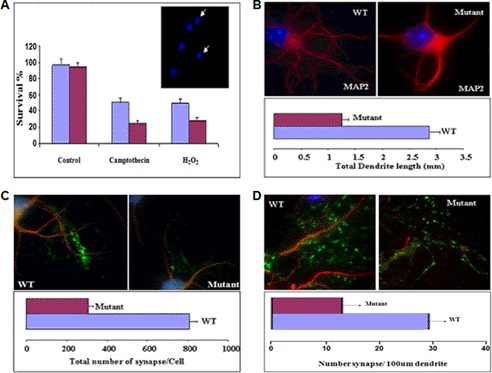
Mutation of the coiled-coil and C2 domain-containing 1A (CC2D1A) gene, which encodes a C2 domain and DM14 domain-containing protein, has been linked to severe autosomal recessive nonsyndromic mental retardation. Using a mouse model that produces a truncated form of CC2D1A that lacks the C2 domain and three of the four DM14 domains, we show that CC2D1A is important for neuronal differentiation and brain development. CC2D1A mutant neurons are hypersensitive to stress and have a reduced capacity to form dendrites and synapses in culture. At the biochemical level, CC2D1A transduces signals to the cyclic adenosine 3',5'-monophosphate (cAMP)-protein kinase A (PKA) pathway during neuronal cell differentiation. PKA activity is compromised, and the translocation of its catalytic subunit to the nucleus is also defective in CC2D1A mutant cells. Consistently, phosphorylation of the PKA target cAMP-responsive element-binding protein, at serine 133, is nearly abolished in CC2D1A mutant cells. The defects in cAMP/PKA signaling were observed in fibroblast, macrophage, and neuronal primary cells derived from the CC2D1A KO mice. CC2D1A associates with the cAMP-PKA complex following forskolin treatment and accumulates in vesicles or on the plasma membrane in wild-type cells, suggesting that CC2D1A may recruit the PKA complex to the membrane to facilitate signal transduction. Together, our data show that CC2D1A is an important regulator of the cAMP/PKA signaling pathway, which may be the underlying cause for impaired mental function in nonsyndromic mental retardation patients with CC2D1A mutation.
Figures
References
-
- Basel-Vanagaite L. (2007) Genetics of autosomal recessive nonsyndromic mental retardation. Recent advances. Clin. Genet. 72, 167–174 - PubMed
-
- Ropers H. H. (2006) X-linked mental retardation. Many genes for a complex disorder. Curr. Opin. Genet. Dev. 16, 260–269 - PubMed
-
- Raymond F. L., Tarpey P. (2006) The genetics of mental retardation. Hum. Mol. Genet. 15, R110–R116 - PubMed
-
- Basel-Vanagaite L., Attia R., Yahav M., Ferland R. J., Anteki L., Walsh C. A., Olender T., Straussberg R., Magal N., Taub E., Drasinover V., Alkelai A., Bercovich D., Rechavi G., Simon A. J., Shohat M. (2006) The CC2D1A, a member of a new gene family with C2 domains, is involved in autosomal recessive nonsyndromic mental retardation. J. Med. Genet. 43, 203–210 - PMC - PubMed
-
- Kikkawa U., Kishimoto A., Nishizuka Y. (1989) The protein kinase C family. Heterogeneity and its implications. Annu. Rev. Biochem. 58, 31–44 - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
