

Identification of 2 novel ANTXR2 mutations in patients with hyaline fibromatosis syndrome and proposal of a modified grading system
- PMID: 22383261
- PMCID: PMC4264531
- DOI: 10.1002/ajmg.a.35228
Identification of 2 novel ANTXR2 mutations in patients with hyaline fibromatosis syndrome and proposal of a modified grading system
Abstract
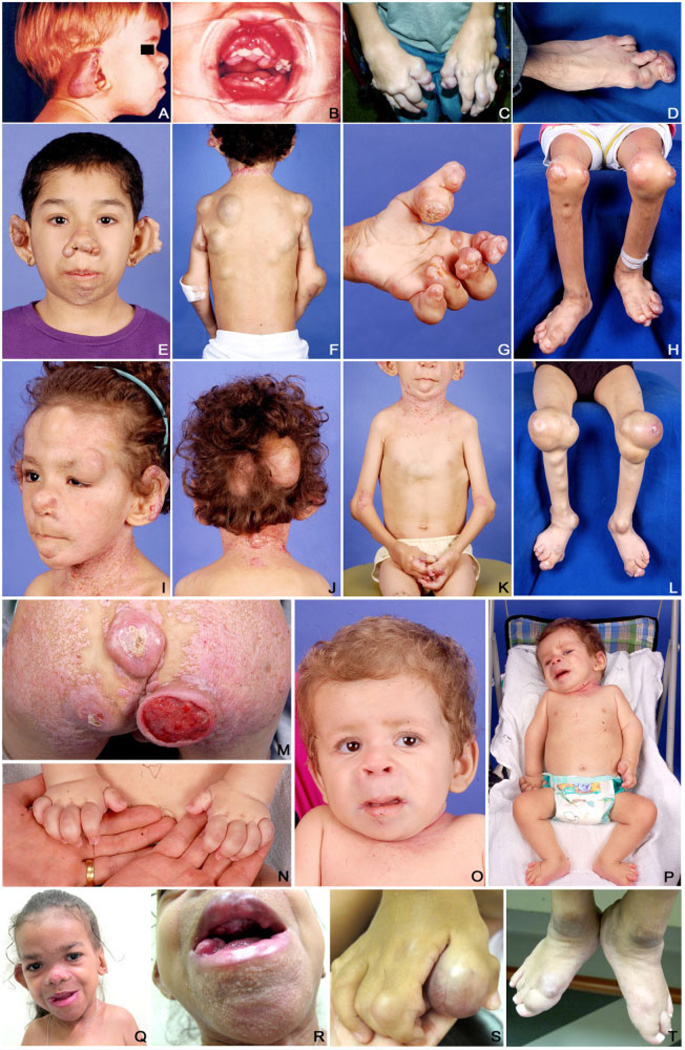
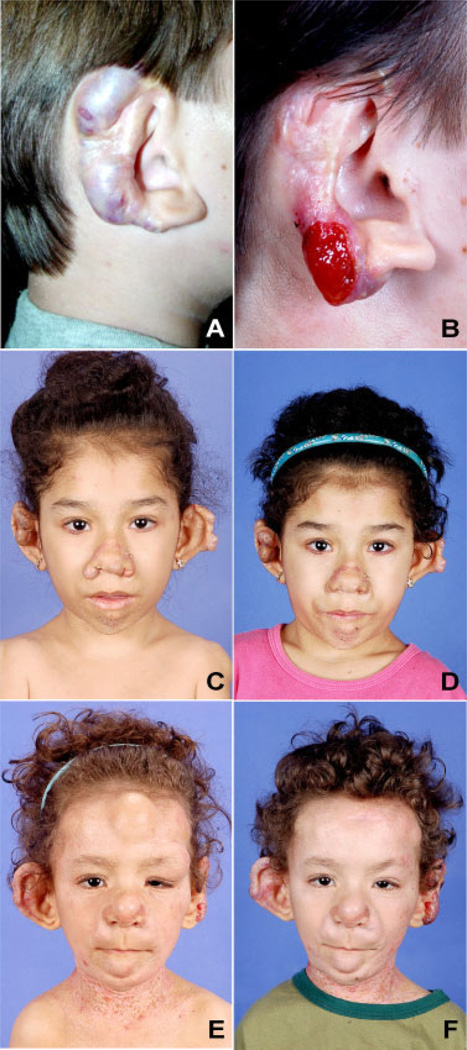
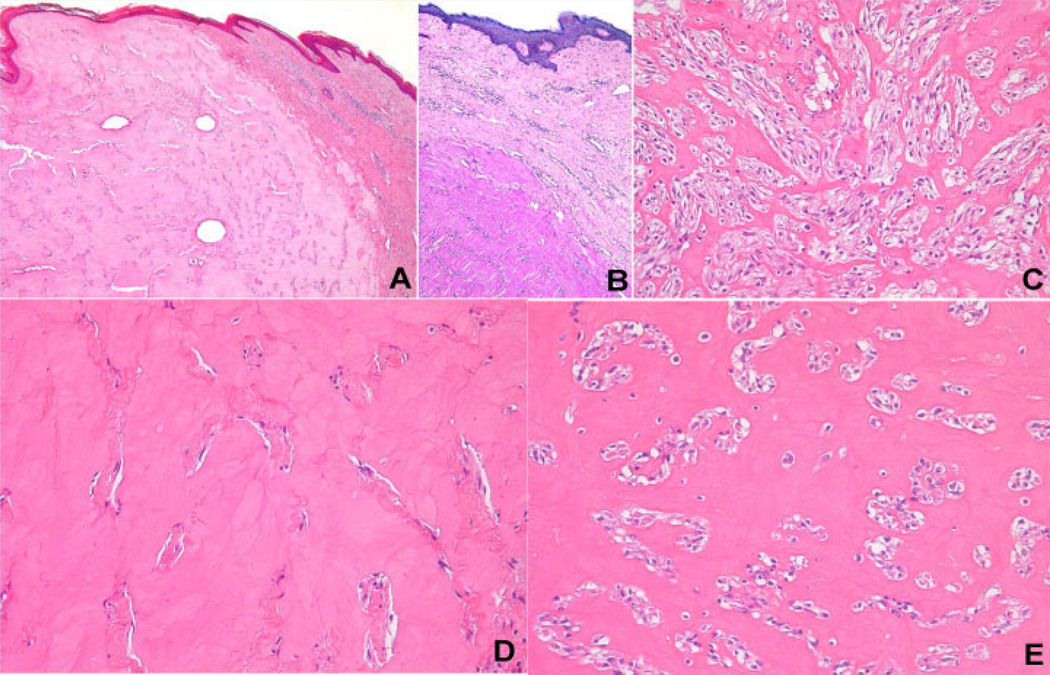
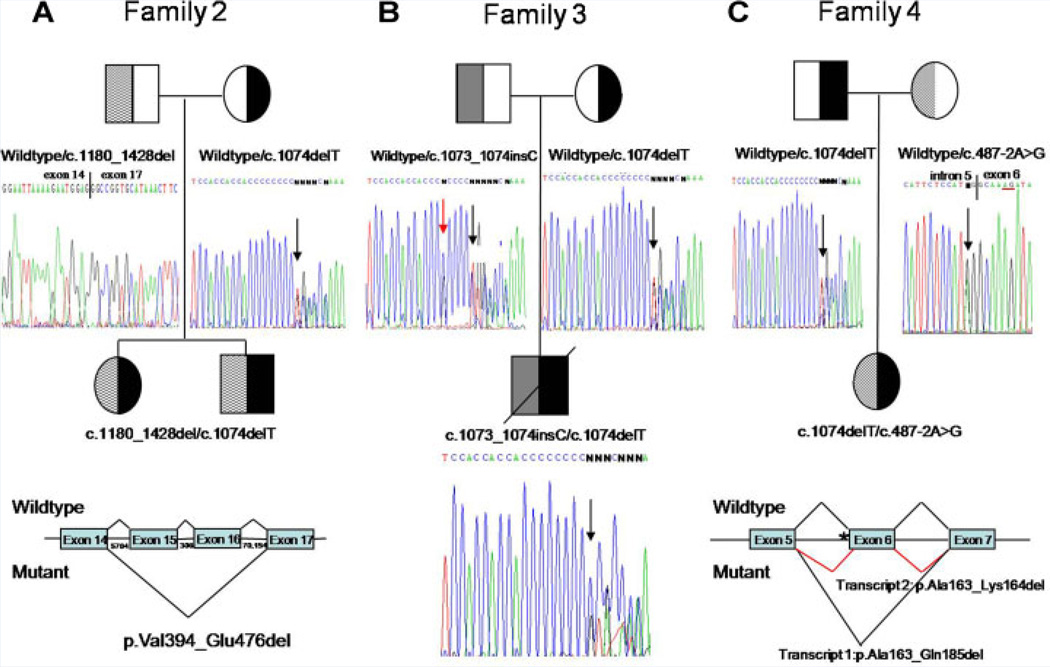
Juvenile hyaline fibromatosis (JHF) and infantile systemic hyalinosis (ISH) are rare, autosomal recessive disorders of the connective tissue caused by mutations in the gene encoding the anthrax toxin receptor 2 protein (ANTXR2) located on chromosome 4q21. Characteristically, these conditions present with overlapping clinical features, such as nodules and/or pearly papules, gingival hyperplasia, flexion contractures of the joints, and osteolytic bone defects. The present report describes a pair of sibs and three other JHF/ISH patients whose diagnoses were based on typical clinical manifestations and confirmed by histopathologic analyses and/or molecular analysis. A comparison of ISH and JHF, additional thoughts about new terminology (hyaline fibromatosis syndrome) and a modified grading system are also included.
Copyright © 2012 Wiley Periodicals, Inc.
Conflict of interest statement
Sources of funding: None/Conflict of interest: None
Figures
References
-
- Antaya RJ, Cajaiba MM, Madri J, Lopez MA, Ramirez MC, Martignetti JA, Reyes-Múgica M. Juvenile hyaline fibromatosis and infantile systemic hyalinosis overlap associated with a novel mutation in capillary morphogenesis protein-2 gene. Am J Dermatopathol. 2007;29:99–103. - PubMed
-
- Brandão FV, Silva CM, Gontijo B, Guedes AC. Juvenile hyaline fibromatosis and infantile systemic hyalinosis. Case for diagnosis. An Bras Dermatol. 2009;84:677–679. - PubMed
-
- Deuquet J, Lausch E, Guex N, Abrami L, Salvi S, Lakkaraju A, Ramirez MC, Martignetti JA, Rokicki D, Bonafe L, Superti-Furga A, van der Goot FG. Hyaline fibromatosis syndrome inducing mutations in the ecto-domain of anthrax toxin receptor 2 can be rescued by proteasome inhibitors. EMBO Mol Med. 2011;3:208–221. - PMC - PubMed
-
- Dhingra M, Amladi S, Savant S, Nayak C. Juvenile hyaline fibromatosis and infantile systemic hyalinosis: Divergent expressions of the same genetic defect? Indian J Dermatol Venereol Leprol. 2008;74:371–374. - PubMed
-
- Dowling O, Difeo A, Ramirez MC, Tukel T, Narla G, Bonafe L, Kayserili H, Yuksel-Apak M, Paller AS, Norton K, Teebi AS, Grum-Tokars V, Martin GS, Davis GE, Glucksman MJ, Martignetti JA. Mutations in capillary morphogenesis gene-2 result in the allelic disorders juvenile hyaline fibromatosis and infantile systemic hyalinosis. Am J Hum Genet. 2003;73:957–966. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
