

Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes
- PMID: 22383525
- PMCID: PMC3340151
- DOI: 10.1074/jbc.M111.288746
Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes
Abstract
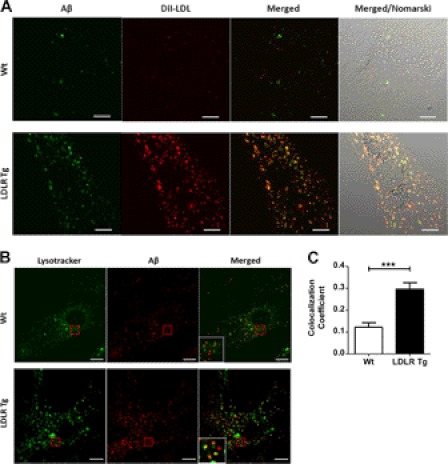
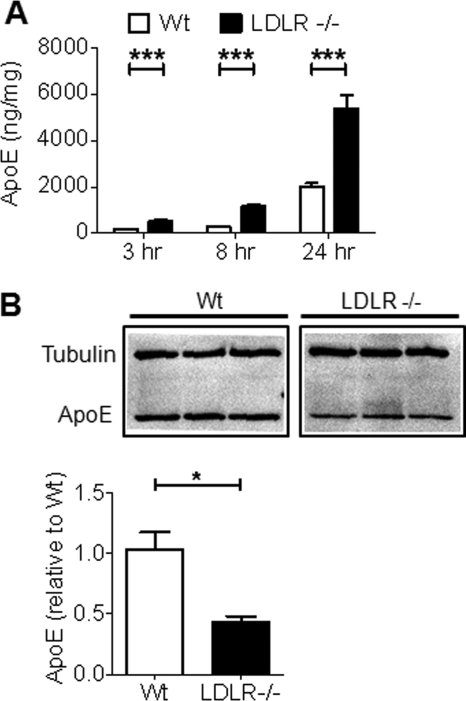
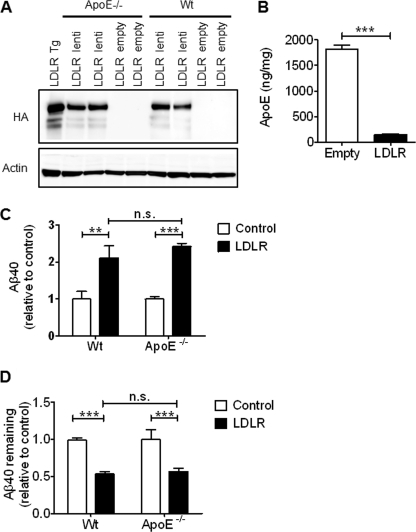
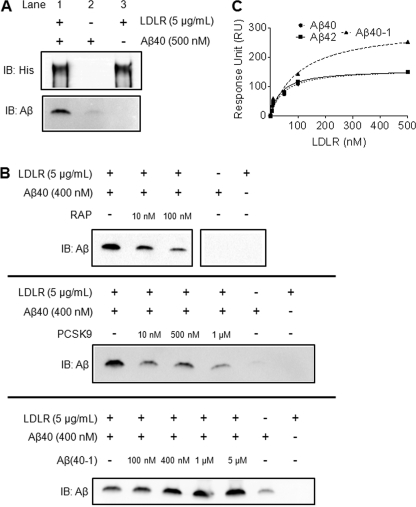
Accumulation of the amyloid β (Aβ) peptide within the brain is hypothesized to be one of the main causes underlying the pathogenic events that occur in Alzheimer disease (AD). Consequently, identifying pathways by which Aβ is cleared from the brain is crucial for better understanding of the disease pathogenesis and developing novel therapeutics. Cellular uptake and degradation by glial cells is one means by which Aβ may be cleared from the brain. In the current study, we demonstrate that modulating levels of the low-density lipoprotein receptor (LDLR), a cell surface receptor that regulates the amount of apolipoprotein E (apoE) in the brain, altered both the uptake and degradation of Aβ by astrocytes. Deletion of LDLR caused a decrease in Aβ uptake, whereas increasing LDLR levels significantly enhanced both the uptake and clearance of Aβ. Increasing LDLR levels also enhanced the cellular degradation of Aβ and facilitated the vesicular transport of Aβ to lysosomes. Despite the fact that LDLR regulated the uptake of apoE by astrocytes, we found that the effect of LDLR on Aβ uptake and clearance occurred in the absence of apoE. Finally, we provide evidence that Aβ can directly bind to LDLR, suggesting that an interaction between LDLR and Aβ could be responsible for LDLR-mediated Aβ uptake. Therefore, these results identify LDLR as a receptor that mediates Aβ uptake and clearance by astrocytes, and provide evidence that increasing glial LDLR levels may promote Aβ degradation within the brain.
Figures




Similar articles
-
The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice.J Biol Chem. 2005 Jul 8;280(27):25754-9. doi: 10.1074/jbc.M502143200. Epub 2005 May 11. J Biol Chem. 2005. PMID: 15888448
-
Apolipoprotein E/Amyloid-β Complex Accumulates in Alzheimer Disease Cortical Synapses via Apolipoprotein E Receptors and Is Enhanced by APOE4.Am J Pathol. 2019 Aug;189(8):1621-1636. doi: 10.1016/j.ajpath.2019.04.010. Epub 2019 May 17. Am J Pathol. 2019. PMID: 31108099 Free PMC article.
-
Amyloid-β protein modulates the perivascular clearance of neuronal apolipoprotein E in mouse models of Alzheimer's disease.J Neural Transm (Vienna). 2011 May;118(5):699-712. doi: 10.1007/s00702-010-0572-7. Epub 2011 Jan 6. J Neural Transm (Vienna). 2011. PMID: 21210284
-
Functional role of lipoprotein receptors in Alzheimer's disease.Curr Alzheimer Res. 2008 Feb;5(1):15-25. doi: 10.2174/156720508783884675. Curr Alzheimer Res. 2008. PMID: 18288927 Review.
-
LRP in amyloid-beta production and metabolism.Ann N Y Acad Sci. 2006 Nov;1086:35-53. doi: 10.1196/annals.1377.005. Ann N Y Acad Sci. 2006. PMID: 17185504 Review.
Cited by
-
Emerging Alzheimer's disease therapeutics: promising insights from lipid metabolism and microglia-focused interventions.Front Aging Neurosci. 2023 Oct 25;15:1259012. doi: 10.3389/fnagi.2023.1259012. eCollection 2023. Front Aging Neurosci. 2023. PMID: 38020773 Free PMC article. Review.
-
Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Aβ clearance in a mouse model of β-amyloidosis.Proc Natl Acad Sci U S A. 2012 Sep 18;109(38):15502-7. doi: 10.1073/pnas.1206446109. Epub 2012 Aug 27. Proc Natl Acad Sci U S A. 2012. PMID: 22927427 Free PMC article.
-
Amyloid-β in Alzheimer's disease - front and centre after all?Neuronal Signal. 2023 Jan 6;7(1):NS20220086. doi: 10.1042/NS20220086. eCollection 2023 Mar. Neuronal Signal. 2023. PMID: 36687366 Free PMC article. Review.
-
ApoE and Aβ in Alzheimer's disease: accidental encounters or partners?Neuron. 2014 Feb 19;81(4):740-54. doi: 10.1016/j.neuron.2014.01.045. Neuron. 2014. PMID: 24559670 Free PMC article. Review.
-
Enhancing astrocytic lysosome biogenesis facilitates Aβ clearance and attenuates amyloid plaque pathogenesis.J Neurosci. 2014 Jul 16;34(29):9607-20. doi: 10.1523/JNEUROSCI.3788-13.2014. J Neurosci. 2014. PMID: 25031402 Free PMC article.
References
-
- Hardy J., Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer disease. Progress and problems on the road to therapeutics. Science 297, 353–356 - PubMed
-
- De Strooper B. (2010) Proteases and proteolysis in Alzheimer disease. A multifactorial view on the disease process. Physiol. Rev. 90, 465–494 - PubMed
-
- Hardy J. (2006) A hundred years of Alzheimer disease research. Neuron 52, 3–13 - PubMed
-
- Selkoe D. J. (2001) Clearing the brain's amyloid cobwebs. Neuron 32, 177–180 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
