

Ten years of pathway analysis: current approaches and outstanding challenges
- PMID: 22383865
- PMCID: PMC3285573
- DOI: 10.1371/journal.pcbi.1002375
Ten years of pathway analysis: current approaches and outstanding challenges
Abstract
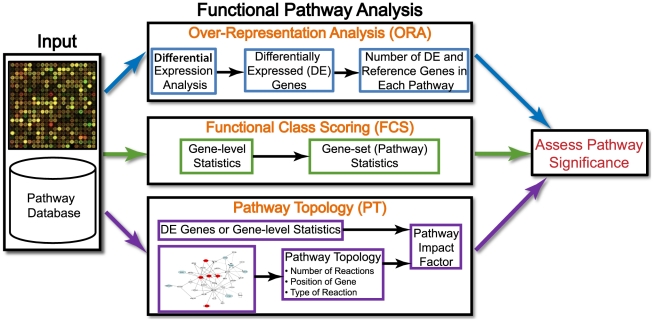
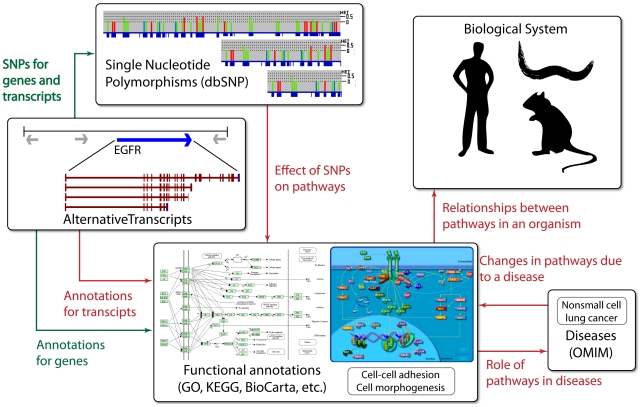
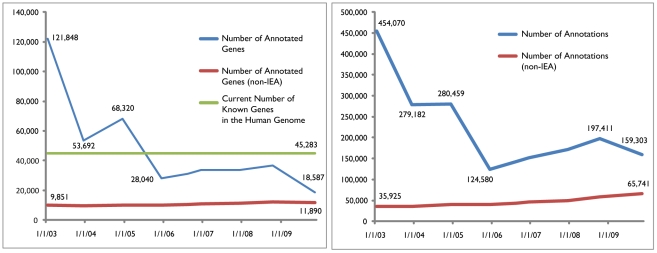
Pathway analysis has become the first choice for gaining insight into the underlying biology of differentially expressed genes and proteins, as it reduces complexity and has increased explanatory power. We discuss the evolution of knowledge base-driven pathway analysis over its first decade, distinctly divided into three generations. We also discuss the limitations that are specific to each generation, and how they are addressed by successive generations of methods. We identify a number of annotation challenges that must be addressed to enable development of the next generation of pathway analysis methods. Furthermore, we identify a number of methodological challenges that the next generation of methods must tackle to take advantage of the technological advances in genomics and proteomics in order to improve specificity, sensitivity, and relevance of pathway analysis.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Goeman JJ, Buhlmann P. Analyzing gene expression data in terms of gene sets: methodological issues. Bioinformatics. 2007;23:980–987. - PubMed
-
- Khatri P, Drăghici S, Ostermeier GC, Krawetz SA. Profiling gene expression using Onto-Express. Genomics. 2002;79:266–270. - PubMed
-
- Drăghici S, Khatri P, Martins RP, Ostermeier GC, Krawetz SA. Global functional profiling of gene expression. Genomics. 2003;81:98–104. - PubMed
