

Structural consensus among antibodies defines the antigen binding site
- PMID: 22383868
- PMCID: PMC3285572
- DOI: 10.1371/journal.pcbi.1002388
Structural consensus among antibodies defines the antigen binding site
Abstract
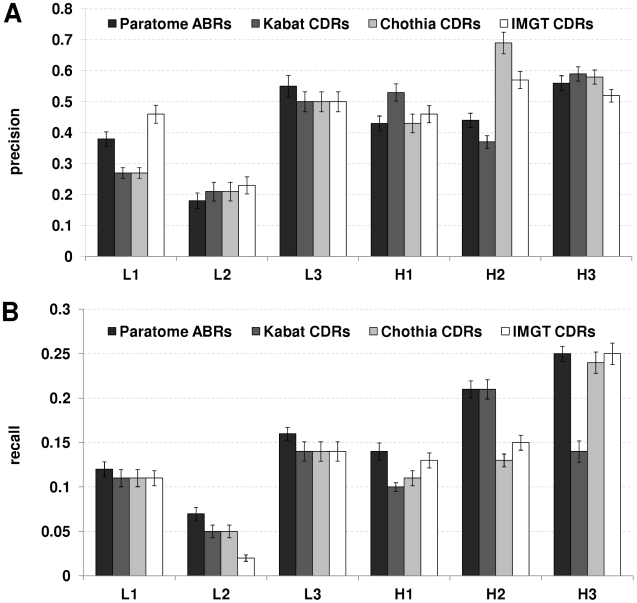
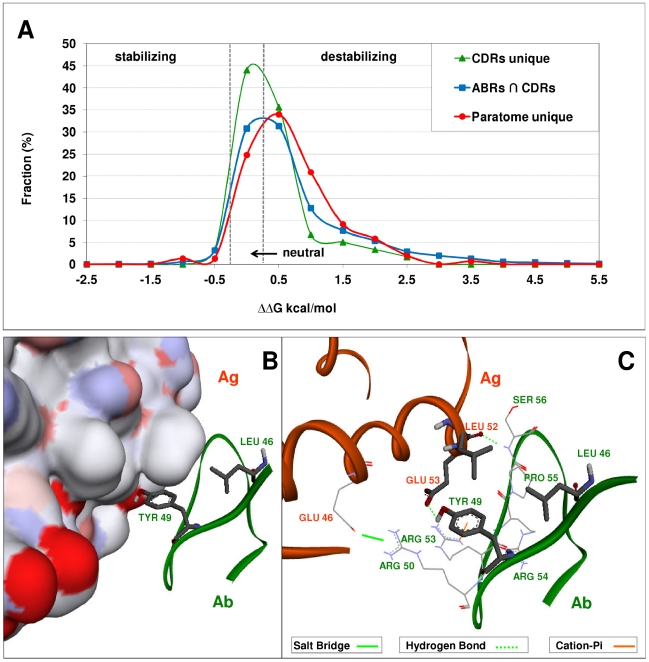
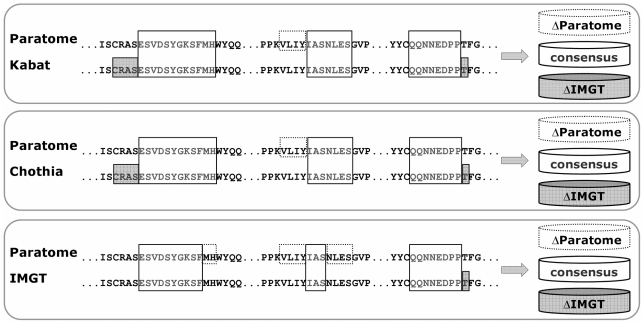
The Complementarity Determining Regions (CDRs) of antibodies are assumed to account for the antigen recognition and binding and thus to contain also the antigen binding site. CDRs are typically discerned by searching for regions that are most different, in sequence or in structure, between different antibodies. Here, we show that ~20% of the antibody residues that actually bind the antigen fall outside the CDRs. However, virtually all antigen binding residues lie in regions of structural consensus across antibodies. Furthermore, we show that these regions of structural consensus which cover the antigen binding site are identifiable from the sequence of the antibody. Analyzing the predicted contribution of antigen binding residues to the stability of the antibody-antigen complex, we show that residues that fall outside of the traditionally defined CDRs are at least as important to antigen binding as residues within the CDRs, and in some cases, they are even more important energetically. Furthermore, antigen binding residues that fall outside of the structural consensus regions but within traditionally defined CDRs show a marginal energetic contribution to antigen binding. These findings allow for systematic and comprehensive identification of antigen binding sites, which can improve the understanding of antigenic interactions and may be useful in antibody engineering and B-cell epitope identification.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
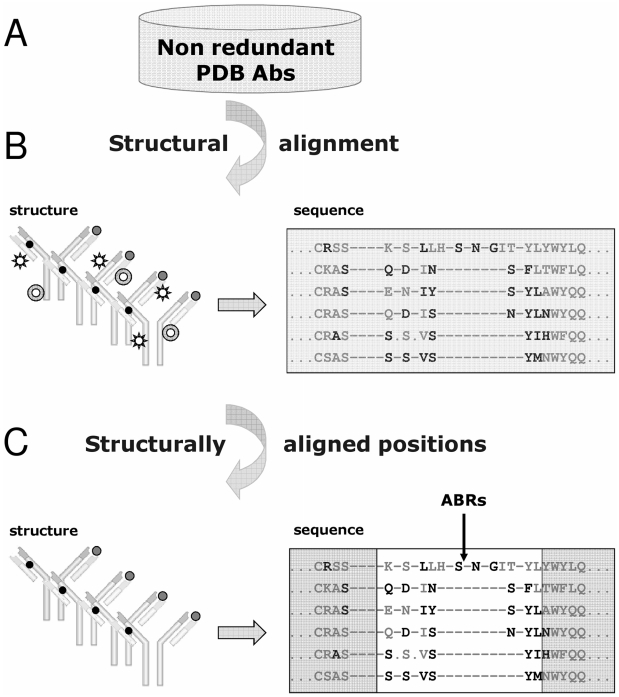
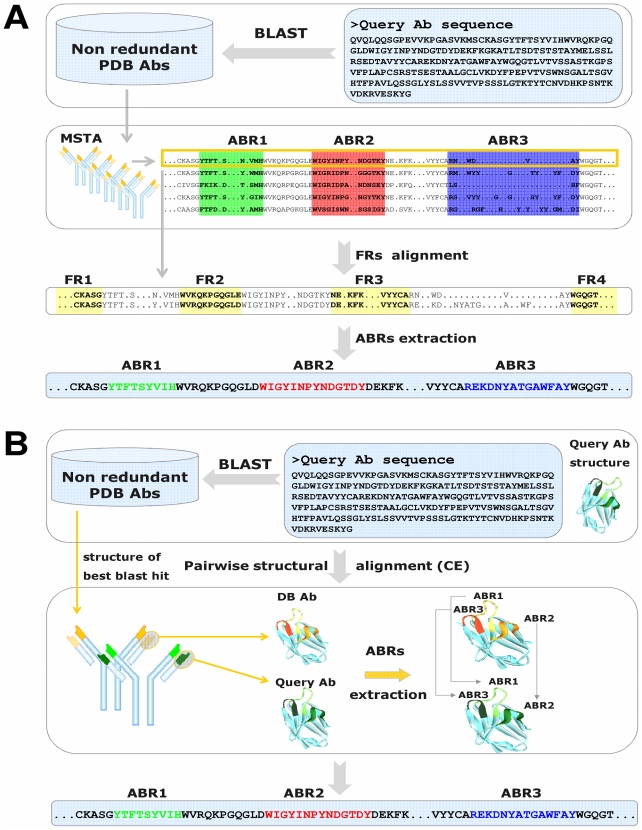
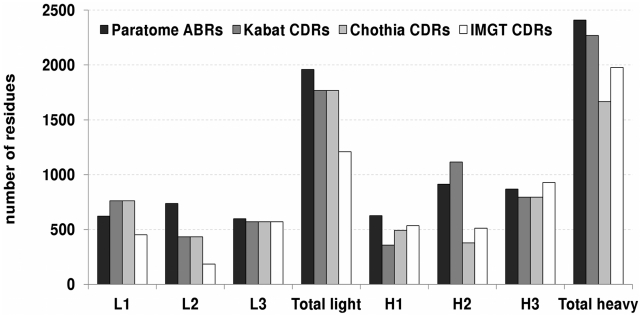
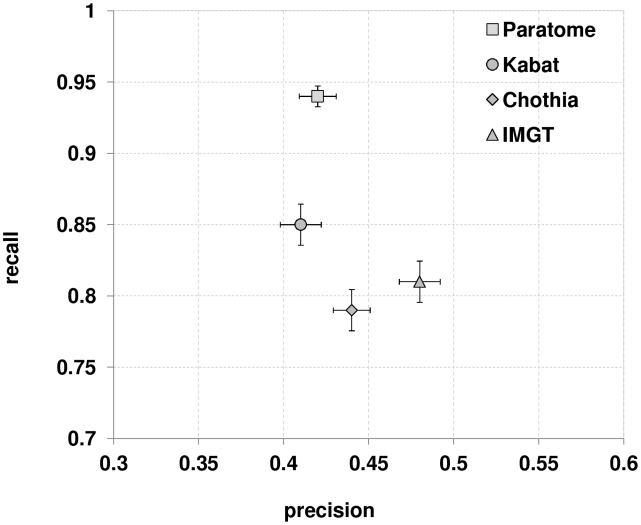
Similar articles
-
Automated identification of complementarity determining regions (CDRs) reveals peculiar characteristics of CDRs and B cell epitopes.J Immunol. 2008 Nov 1;181(9):6230-5. doi: 10.4049/jimmunol.181.9.6230. J Immunol. 2008. PMID: 18941213
-
Paratome: an online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure.Nucleic Acids Res. 2012 Jul;40(Web Server issue):W521-4. doi: 10.1093/nar/gks480. Epub 2012 Jun 6. Nucleic Acids Res. 2012. PMID: 22675071 Free PMC article.
-
Antigen-Antibody Complexes.Subcell Biochem. 2020;94:465-497. doi: 10.1007/978-3-030-41769-7_19. Subcell Biochem. 2020. PMID: 32189312 Review.
-
A computational approach for studying antibody-antigen interactions without prior structural information: the anti-testosterone binding antibody as a case study.Proteins. 2017 Feb;85(2):322-331. doi: 10.1002/prot.25226. Epub 2016 Dec 26. Proteins. 2017. PMID: 27936519
-
Specificity, polyspecificity, and heterospecificity of antibody-antigen recognition.J Mol Recognit. 2014 Nov;27(11):627-39. doi: 10.1002/jmr.2394. J Mol Recognit. 2014. PMID: 25277087 Review.
Cited by
-
Humanization of the antigen-recognition domain does not impinge on the antigen-binding, cytokine secretion, and antitumor reactivity of humanized nanobody-based CD19-redirected CAR-T cells.J Transl Med. 2024 Jul 25;22(1):679. doi: 10.1186/s12967-024-05461-8. J Transl Med. 2024. PMID: 39054481 Free PMC article.
-
Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes.Proc Natl Acad Sci U S A. 2015 Aug 11;112(32):9967-72. doi: 10.1073/pnas.1511996112. Epub 2015 Jul 27. Proc Natl Acad Sci U S A. 2015. PMID: 26216968 Free PMC article.
-
Antibody Structure and Function: The Basis for Engineering Therapeutics.Antibodies (Basel). 2019 Dec 3;8(4):55. doi: 10.3390/antib8040055. Antibodies (Basel). 2019. PMID: 31816964 Free PMC article. Review.
-
Analysis of nanobody paratopes reveals greater diversity than classical antibodies.Protein Eng Des Sel. 2018 Jul 1;31(7-8):267-275. doi: 10.1093/protein/gzy017. Protein Eng Des Sel. 2018. PMID: 30053276 Free PMC article.
-
Characterization of the naive murine antibody repertoire using unamplified high-throughput sequencing.PLoS One. 2018 Jan 10;13(1):e0190982. doi: 10.1371/journal.pone.0190982. eCollection 2018. PLoS One. 2018. PMID: 29320559 Free PMC article.
References
-
- Crameri A, Cwirla S, Stemmer WP. Construction and evolution of antibody-phage libraries by DNA shuffling. Nat Med. 1996;2:100–102. - PubMed
-
- Figini M, Marks JD, Winter G, Griffiths AD. In vitro assembly of repertoires of antibody chains on the surface of phage by renaturation. J Mol Biol. 1994;239:68–78. - PubMed
-
- Hawkins RE, Russell SJ, Winter G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J Mol Biol. 1992;226:889–896. - PubMed
-
- Lou J, Marks JD. Affinity Maturation by Chain Shuffling and Site Directed Mutagenesis. In: Konterman R, Dubel S, editors. Antibody Engineering. New York: Springer; 2010. pp. 377–396.
-
- Almagro JC. Identification of differences in the specificity-determining residues of antibodies that recognize antigens of different size: implications for the rational design of antibody repertoires. J Mol Recognit. 2004;17:132–143. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
