

Loss of Tgif function causes holoprosencephaly by disrupting the SHH signaling pathway
- PMID: 22383895
- PMCID: PMC3285584
- DOI: 10.1371/journal.pgen.1002524
Loss of Tgif function causes holoprosencephaly by disrupting the SHH signaling pathway
Abstract
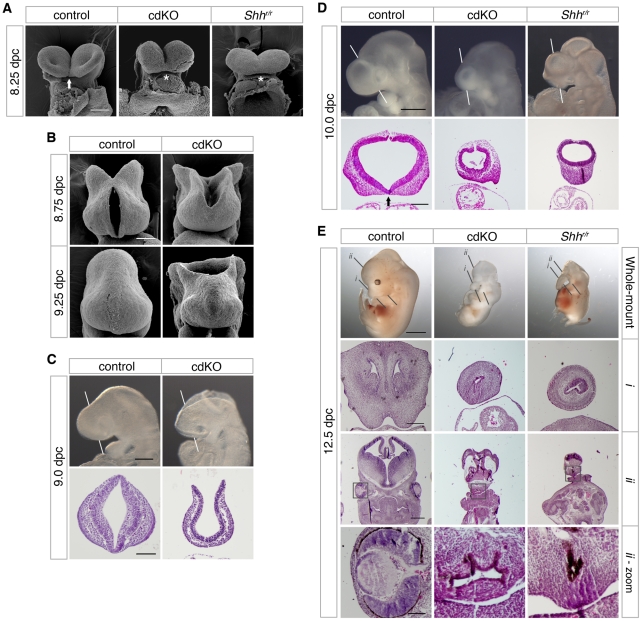
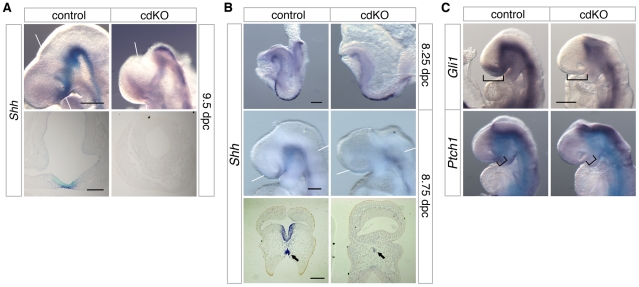
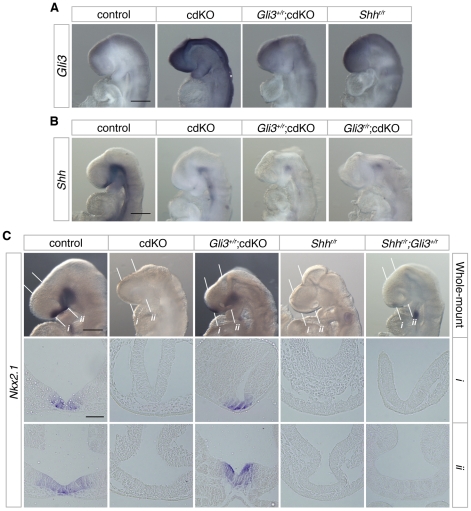
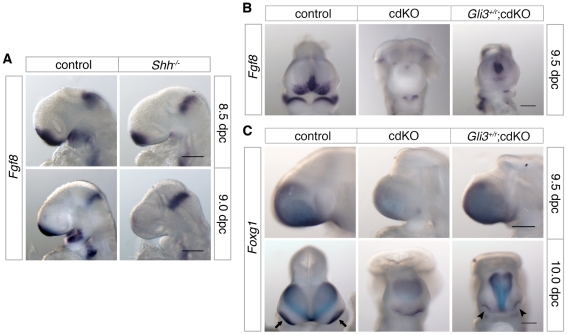
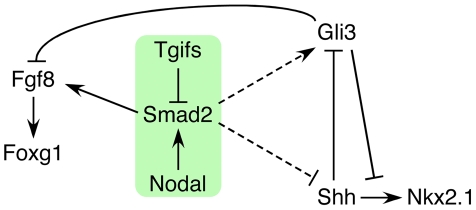
Holoprosencephaly (HPE) is a severe human genetic disease affecting craniofacial development, with an incidence of up to 1/250 human conceptions and 1.3 per 10,000 live births. Mutations in the Sonic Hedgehog (SHH) gene result in HPE in humans and mice, and the Shh pathway is targeted by other mutations that cause HPE. However, at least 12 loci are associated with HPE in humans, suggesting that defects in other pathways contribute to this disease. Although the TGIF1 (TG-interacting factor) gene maps to the HPE4 locus, and heterozygous loss of function TGIF1 mutations are associated with HPE, mouse models have not yet explained how loss of Tgif1 causes HPE. Using a conditional Tgif1 allele, we show that mouse embryos lacking both Tgif1 and the related Tgif2 have HPE-like phenotypes reminiscent of Shh null embryos. Eye and nasal field separation is defective, and forebrain patterning is disrupted in embryos lacking both Tgifs. Early anterior patterning is relatively normal, but expression of Shh is reduced in the forebrain, and Gli3 expression is up-regulated throughout the neural tube. Gli3 acts primarily as an antagonist of Shh function, and the introduction of a heterozygous Gli3 mutation into embryos lacking both Tgif genes partially rescues Shh signaling, nasal field separation, and HPE. Tgif1 and Tgif2 are transcriptional repressors that limit Transforming Growth Factor β/Nodal signaling, and we show that reducing Nodal signaling in embryos lacking both Tgifs reduces the severity of HPE and partially restores the output of Shh signaling. Together, these results support a model in which Tgif function limits Nodal signaling to maintain the appropriate output of the Shh pathway in the forebrain. These data show for the first time that Tgif1 mutation in mouse contributes to HPE pathogenesis and provide evidence that this is due to disruption of the Shh pathway.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures




Similar articles
-
Genetic and Molecular Analyses indicate independent effects of TGIFs on Nodal and Gli3 in neural tube patterning.Eur J Hum Genet. 2017 Feb;25(2):208-215. doi: 10.1038/ejhg.2016.164. Epub 2016 Dec 7. Eur J Hum Genet. 2017. PMID: 27924807 Free PMC article.
-
Functions of TGIF homeodomain proteins and their roles in normal brain development and holoprosencephaly.Am J Med Genet C Semin Med Genet. 2018 Jun;178(2):128-139. doi: 10.1002/ajmg.c.31612. Epub 2018 May 11. Am J Med Genet C Semin Med Genet. 2018. PMID: 29749689 Free PMC article.
-
Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination.Nat Genet. 2000 Jun;25(2):205-8. doi: 10.1038/76074. Nat Genet. 2000. PMID: 10835638
-
Mouse models of holoprosencephaly.Curr Opin Neurol. 2003 Apr;16(2):135-41. doi: 10.1097/01.wco.0000063761.15877.40. Curr Opin Neurol. 2003. PMID: 12644739 Review.
-
Modeling the complex etiology of holoprosencephaly in mice.Am J Med Genet C Semin Med Genet. 2018 Jun;178(2):140-150. doi: 10.1002/ajmg.c.31611. Epub 2018 May 11. Am J Med Genet C Semin Med Genet. 2018. PMID: 29749693 Free PMC article. Review.
Cited by
-
Genetic similarity between cancers and comorbid Mendelian diseases identifies candidate driver genes.Nat Commun. 2015 Apr 30;6:7033. doi: 10.1038/ncomms8033. Nat Commun. 2015. PMID: 25926297 Free PMC article.
-
A multi-resource data integration approach: identification of candidate genes regulating cell proliferation during neocortical development.Front Neurosci. 2014 Aug 21;8:257. doi: 10.3389/fnins.2014.00257. eCollection 2014. Front Neurosci. 2014. PMID: 25191221 Free PMC article.
-
Holoprosencephaly: signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans.Wiley Interdiscip Rev Dev Biol. 2015 Jan-Feb;4(1):17-32. doi: 10.1002/wdev.161. Epub 2014 Oct 22. Wiley Interdiscip Rev Dev Biol. 2015. PMID: 25339593 Free PMC article. Review.
-
Identification of conserved skeletal enhancers associated with craniosynostosis risk genes.Hum Mol Genet. 2024 May 4;33(10):837-849. doi: 10.1093/hmg/ddad182. Hum Mol Genet. 2024. PMID: 37883470 Free PMC article.
-
Posttranslational Modifications in Conserved Transcription Factors: A Survey of the TALE-Homeodomain Superclass in Human and Mouse.Front Cell Dev Biol. 2021 Mar 9;9:648765. doi: 10.3389/fcell.2021.648765. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 33768097 Free PMC article. Review.
References
-
- Muenke M, Beachy PA. C.R. S, A.L. B, W.S. S, D. V, B. C, et al., editors. Holoprosencephaly. The metabolic and molecular bases of inherited disease: McGraw-Hill. 2001. pp. 6203–6230.
-
- Leoncini E, Baranello G, Orioli IM, Anneren G, Bakker M, et al. Frequency of holoprosencephaly in the International Clearinghouse Birth Defects Surveillance Systems: searching for population variations. Birth Defects Res A Clin Mol Teratol. 2008;82:585–591. - PubMed
-
- Matsunaga E, Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology. 1977;16:261–272. - PubMed
-
- Roach E, Demyer W, Conneally PM, Palmer C, Merritt AD. Holoprosencephaly: birth data, genetic and demographic analyses of 30 families. Birth Defects Orig Artic Ser. 1975;11:294–313. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
