

Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735
- PMID: 22384041
- PMCID: PMC3288042
- DOI: 10.1371/journal.pone.0031600
Interleukin-1 stimulates ADAM17 through a mechanism independent of its cytoplasmic domain or phosphorylation at threonine 735
Abstract
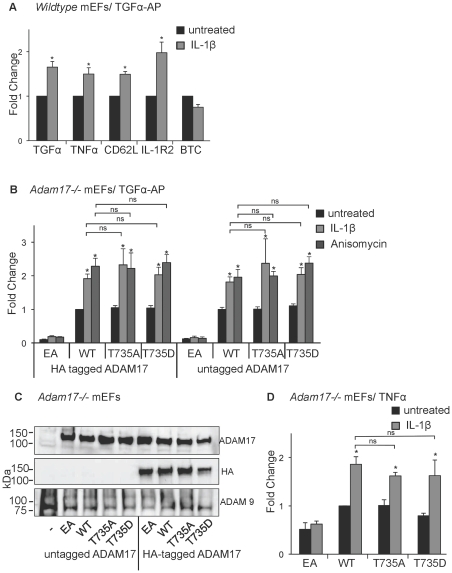
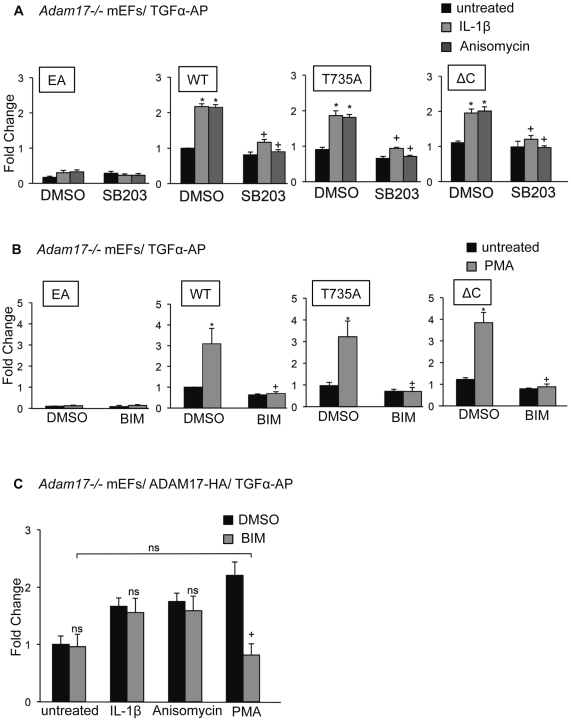
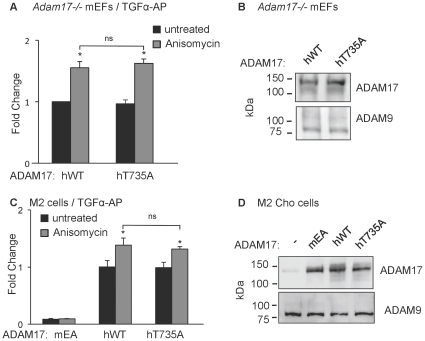
ADAM17 (a disintegrin and metalloproteinase) is a membrane-anchored metalloproteinase that regulates the release of EGFR-ligands, TNFα and other membrane proteins from cells. ADAM17 can be rapidly activated by a variety of signaling pathways, yet little is known about the underlying mechanism. Several studies have demonstrated that the cytoplasmic domain of ADAM17 is not required for its rapid activation by a variety of stimuli, including phorbol esters, tyrosine kinases and some G-protein coupled receptors. However, phosphorylation of cytoplasmic residue T735 was recently reported as a crucial step for activation of ADAM17 by IL-1β and by the p38 MAP-kinase pathway. One possible mechanism to reconcile these results would be that T735 has an inhibitory role and that it must be phosphorylated as a pre-requisite for the activation of ADAM17, which would then proceed via a mechanism that is independent of its cytoplasmic domain. To test this hypothesis, we performed rescue experiments of Adam17-/- cells with wild type and mutant forms of ADAM17. However, these experiments showed that an inactivating mutation (T735A) or an activating mutation (T735D) of cytoplasmic residue T735 or the removal of the cytoplasmic domain of ADAM17 did not significantly affect the stimulation of ADAM17 by IL-1β or by activation of MAP-kinase with anisomycin. Moreover, we found that the MAP-kinase inhibitor SB203580 blocked activation of cytoplasmic tail-deficient ADAM17 and of the T735A mutant by IL-1β or by anisomycin, providing further support for a model in which the activation mechanism of ADAM17 does not rely on its cytoplasmic domain or phosphorylation of T735.
Conflict of interest statement
Figures
References
-
- Black R, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, et al. A metalloprotease disintegrin that releases tumour-necrosis factor-α from cells. Nature. 1997;385:729–733. - PubMed
-
- Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, et al. Cutting Edge: TNF-α-Converting Enzyme (TACE/ADAM17) Inactivation in Mouse Myeloid Cells Prevents Lethality from Endotoxin Shock. J Immunol. 2007;179:2686–2689. - PubMed
-
- Moss ML, Jin S-LC, Milla ME, Burkhart W, Cartner HL, et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-recrosis factor-α. Nature. 1997;385:733–736. - PubMed
-
- Bell J, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-α and its receptors by neutrophils and macrophages. J Leukoc Biol. 2007;82 - PubMed
-
- Blobel CP. ADAMs: key players in EGFR-signaling, development and disease. Nat Rev Mol Cell Bio. 2005;6:32–43. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
