

High-Resolution SNP/CGH Microarrays Reveal the Accumulation of Loss of Heterozygosity in Commonly Used Candida albicans Strains
- PMID: 22384363
- PMCID: PMC3276171
- DOI: 10.1534/g3.111.000885
High-Resolution SNP/CGH Microarrays Reveal the Accumulation of Loss of Heterozygosity in Commonly Used Candida albicans Strains
Erratum in
- G3 (Bethesda). 2012 Nov;2(11):1473
Abstract
Phenotypic diversity can arise rapidly through loss of heterozygosity (LOH) or by the acquisition of copy number variations (CNV) spanning whole chromosomes or shorter contiguous chromosome segments. In Candida albicans, a heterozygous diploid yeast pathogen with no known meiotic cycle, homozygosis and aneuploidy alter clinical characteristics, including drug resistance. Here, we developed a high-resolution microarray that simultaneously detects ∼39,000 single nucleotide polymorphism (SNP) alleles and ∼20,000 copy number variation loci across the C. albicans genome. An important feature of the array analysis is a computational pipeline that determines SNP allele ratios based upon chromosome copy number. Using the array and analysis tools, we constructed a haplotype map (hapmap) of strain SC5314 to assign SNP alleles to specific homologs, and we used it to follow the acquisition of loss of heterozygosity (LOH) and copy number changes in a series of derived laboratory strains. This high-resolution SNP/CGH microarray and the associated hapmap facilitated the phasing of alleles in lab strains and revealed detrimental genome changes that arose frequently during molecular manipulations of laboratory strains. Furthermore, it provided a useful tool for rapid, high-resolution, and cost-effective characterization of changes in allele diversity as well as changes in chromosome copy number in new C. albicans isolates.
Keywords: aneuploidy; comparative genome hybridization; genome profiling; haplotype mapping; loss of heterozygosity; single nucleotide polymorphisms.
Figures
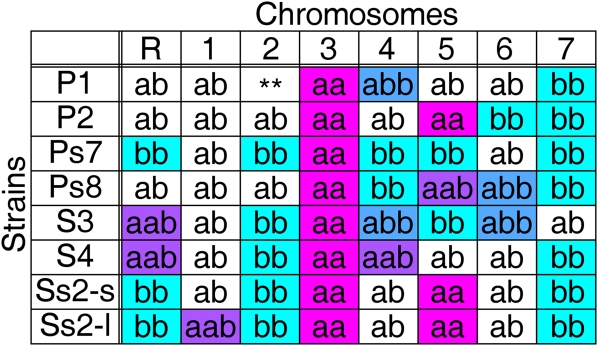
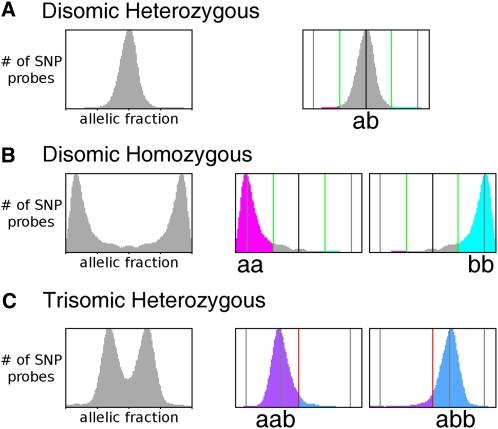

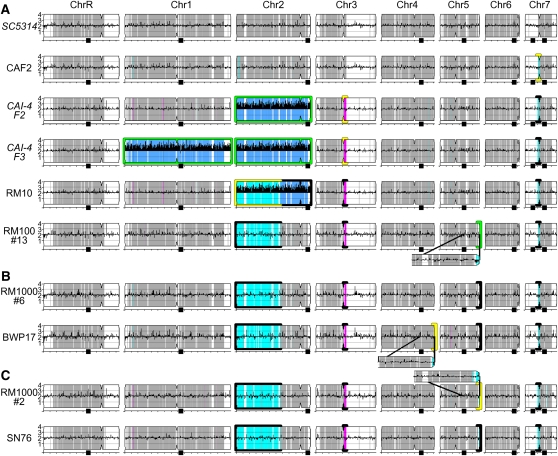
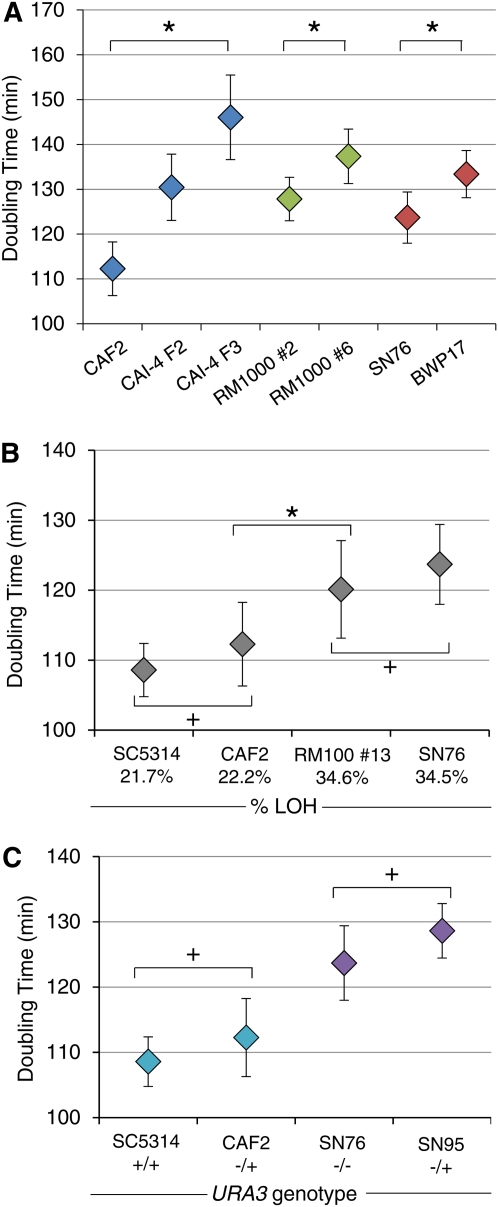
References
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous