

The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke
- PMID: 22388960
- PMCID: PMC3314139
- DOI: 10.1038/nn.3059
The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke
Abstract
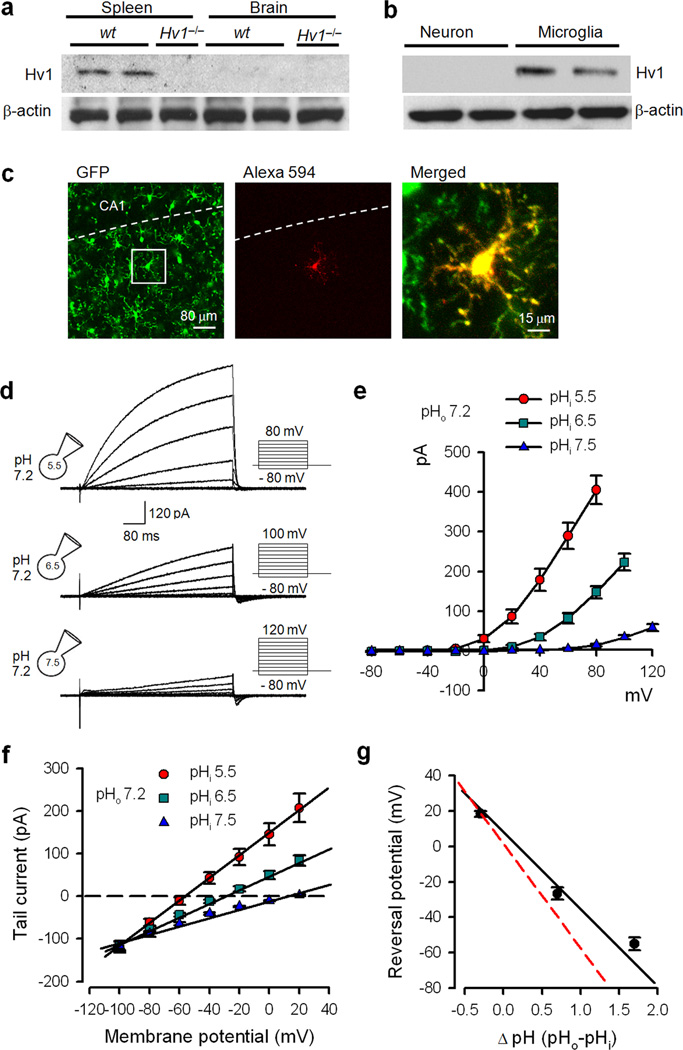
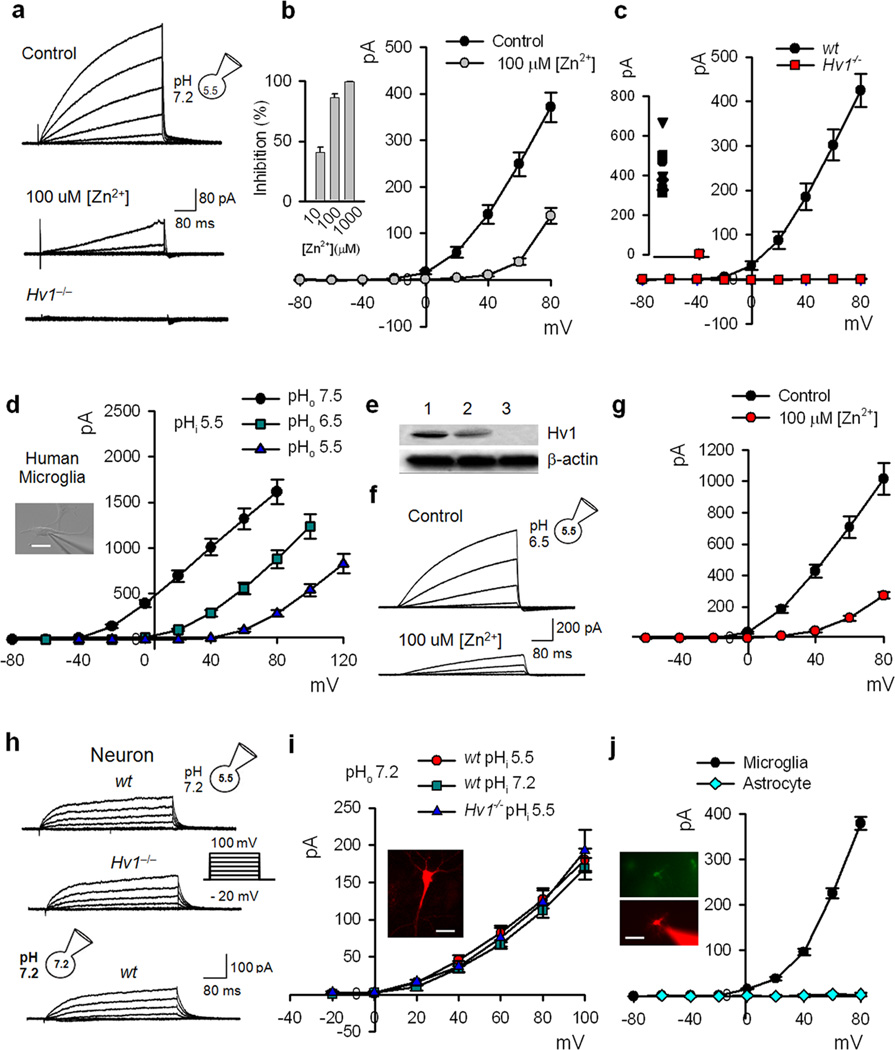
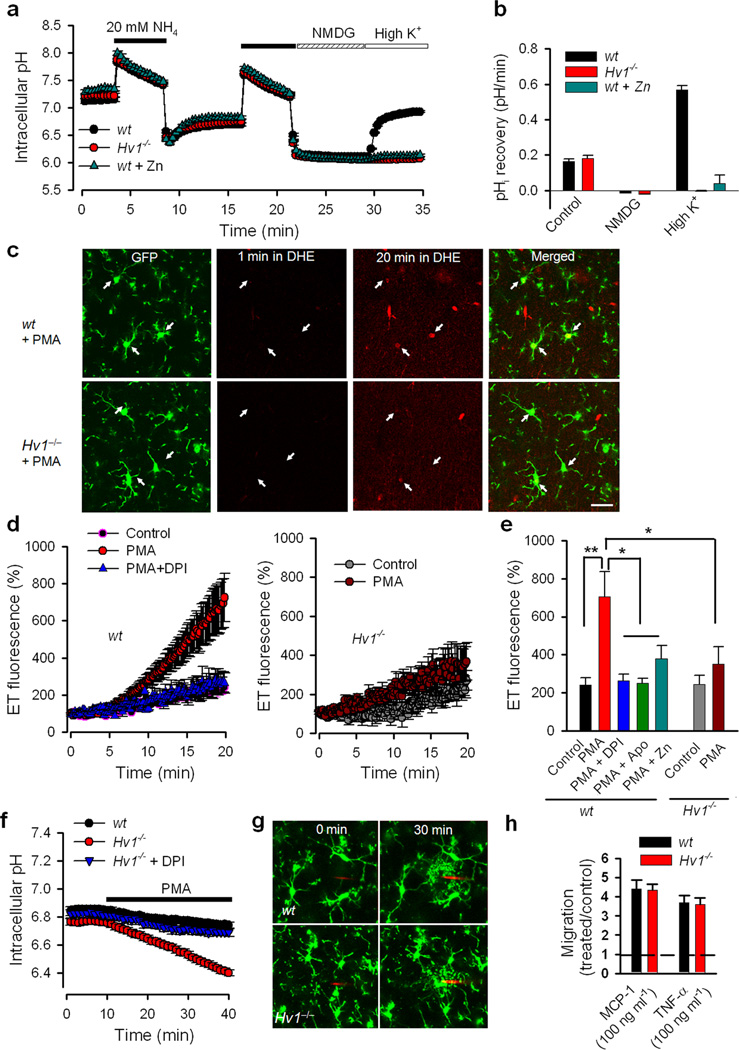
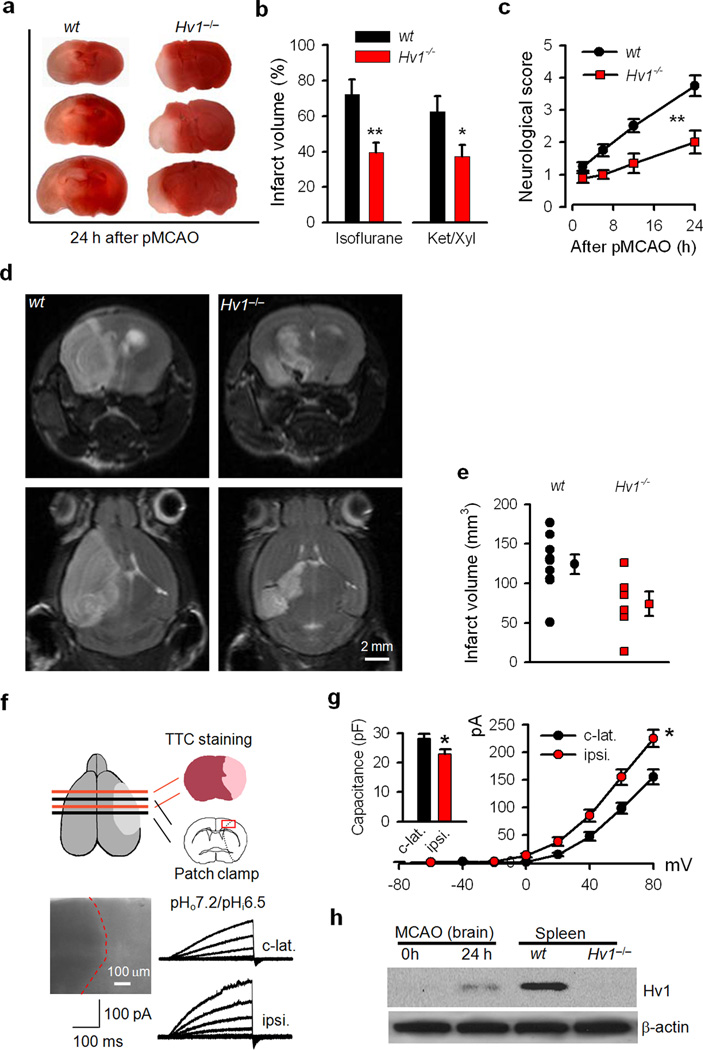
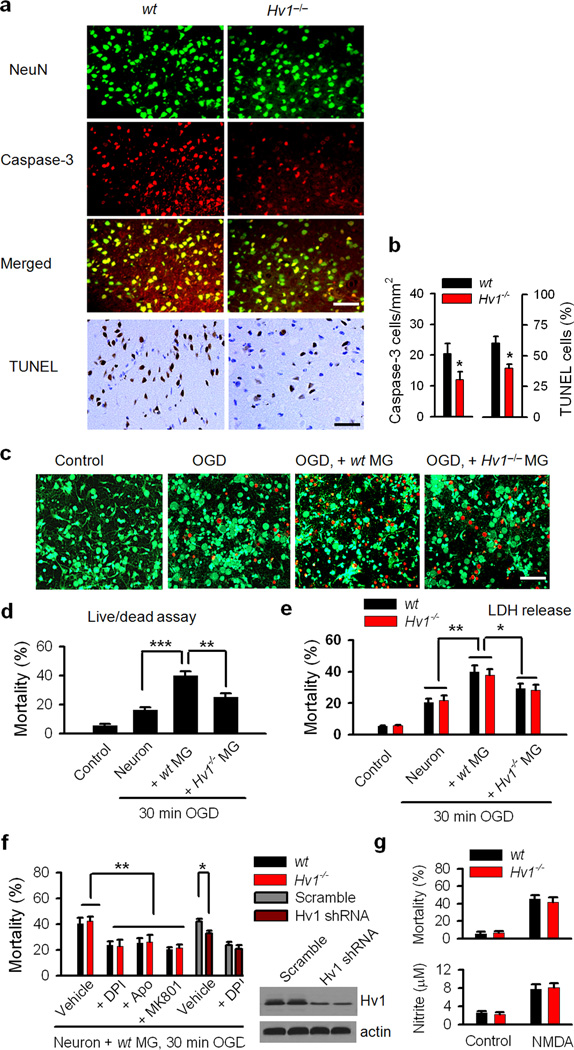
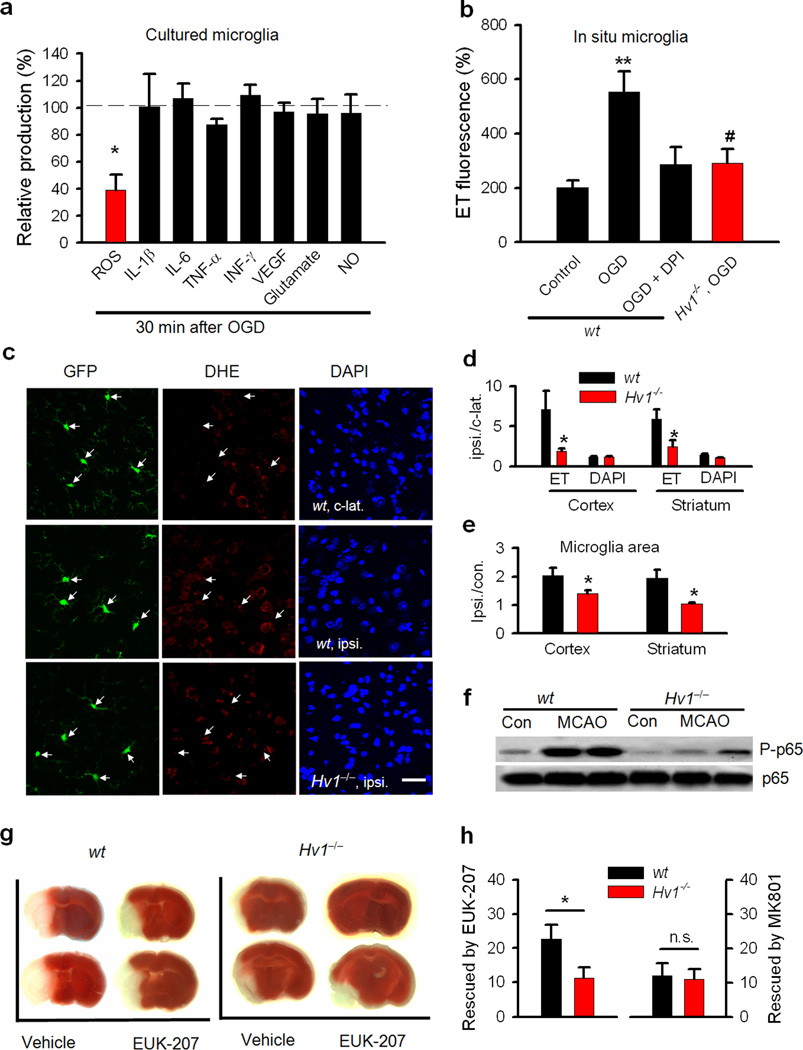
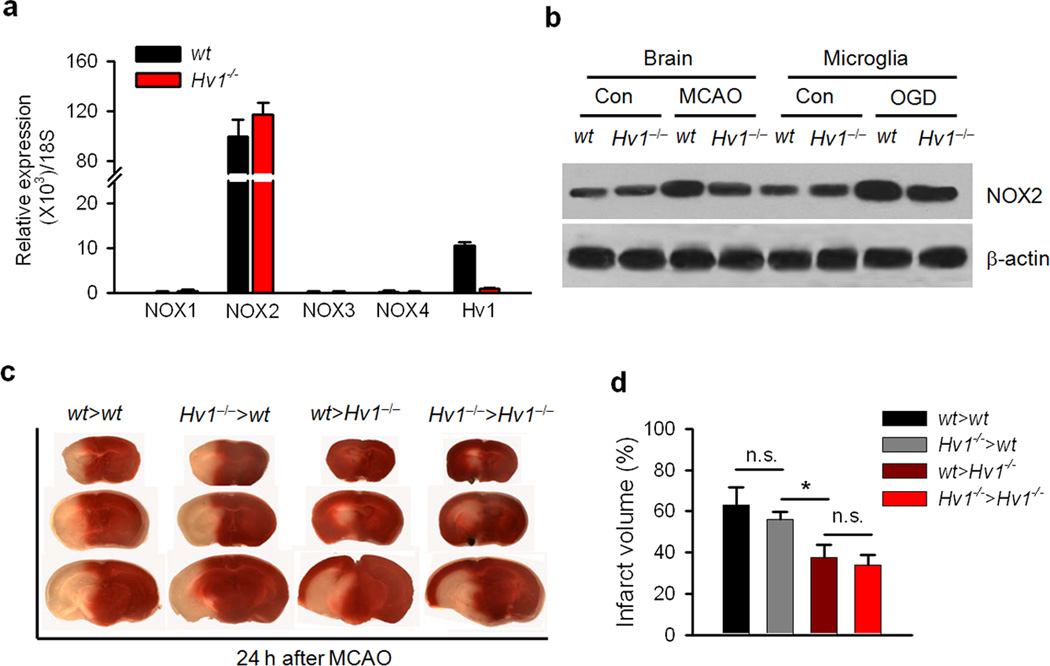
Phagocytic cell NADPH oxidase (NOX) generates reactive oxygen species (ROS) as part of innate immunity. Unfortunately, ischemia can also induce this pathway and inflict damage on native cells. The voltage-gated proton channel Hv1 enables NOX function by compensating cellular loss of electrons with protons. Accordingly, we investigated whether NOX-mediated brain damage in stroke can be inhibited by suppression of Hv1. We found that mouse and human brain microglia, but not neurons or astrocytes, expressed large Hv1-mediated currents. Hv1 was required for NOX-dependent ROS generation in brain microglia in situ and in vivo. Mice lacking Hv1 were protected from NOX-mediated neuronal death and brain damage 24 h after stroke. These results indicate that Hv1-dependent ROS production is responsible for a substantial fraction of brain damage at early time points after ischemic stroke and provide a rationale for Hv1 as a therapeutic target for the treatment of ischemic stroke.
Figures
References
-
- Nathan C, Ding A. SnapShot: Reactive Oxygen Intermediates (ROI) Cell. 2010;140:951–951. e952. - PubMed
-
- Steinhubl SR. Why have antioxidants failed in clinical trials? Am J Cardiol. 2008;101:14D–19D. - PubMed
-
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. - PubMed
-
- Dringen R. Oxidative and antioxidative potential of brain microglial cells. Antioxid Redox Signal. 2005;7:1223–1233. - PubMed
-
- Walder CE, et al. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
