

Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis
- PMID: 22391557
- PMCID: PMC3371709
- DOI: 10.1101/gr.134106.111
Transcriptome survey reveals increased complexity of the alternative splicing landscape in Arabidopsis
Abstract
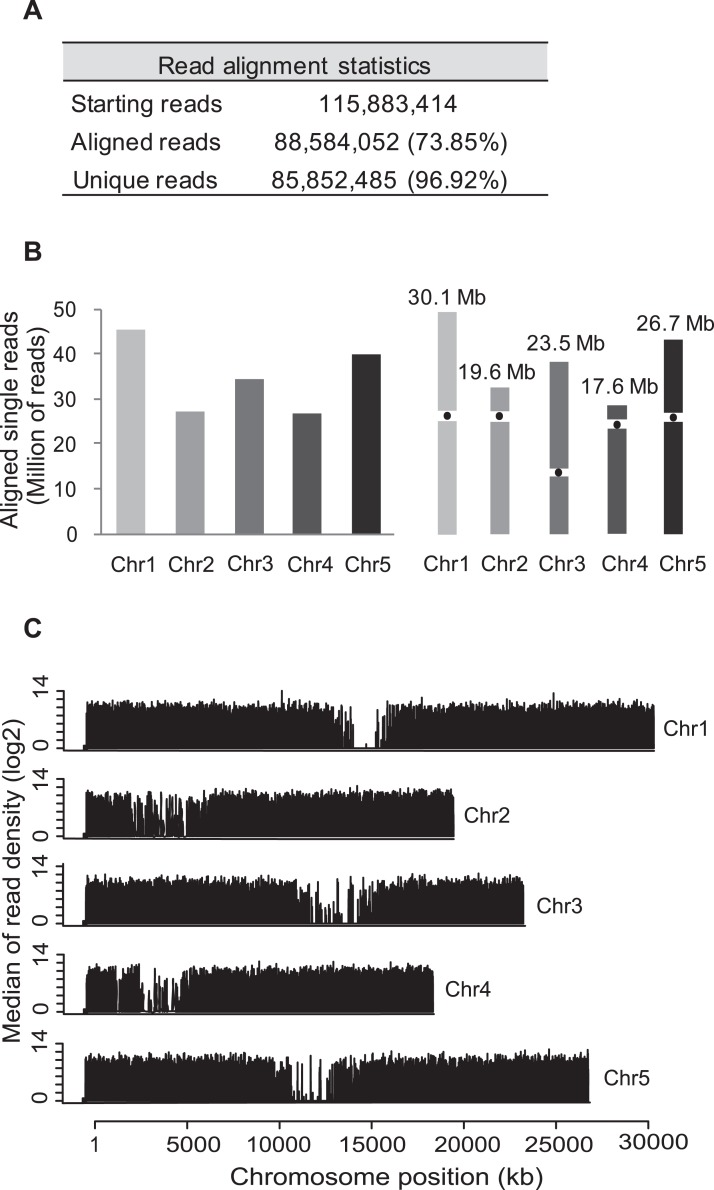
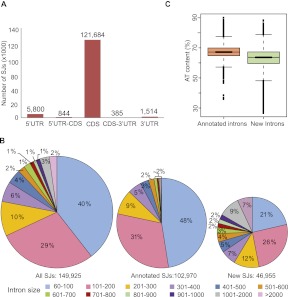
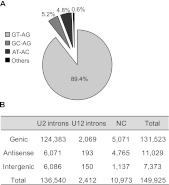
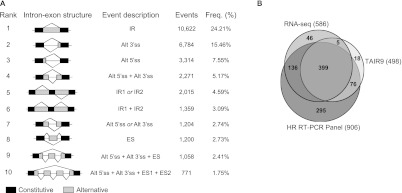
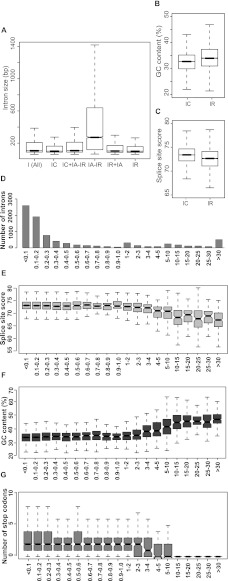
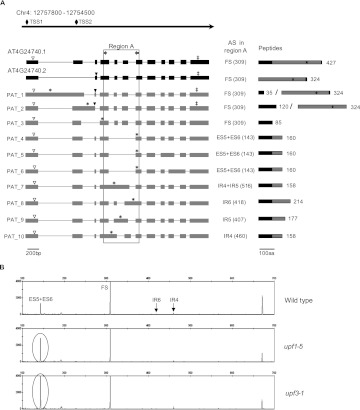
Alternative splicing (AS) is a key regulatory mechanism that contributes to transcriptome and proteome diversity. As very few genome-wide studies analyzing AS in plants are available, we have performed high-throughput sequencing of a normalized cDNA library which resulted in a high coverage transcriptome map of Arabidopsis. We detect ∼150,000 splice junctions derived mostly from typical plant introns, including an eightfold increase in the number of U12 introns (2069). Around 61% of multiexonic genes are alternatively spliced under normal growth conditions. Moreover, we provide experimental validation of 540 AS transcripts (from 256 genes coding for important regulatory factors) using high-resolution RT-PCR and Sanger sequencing. Intron retention (IR) is the most frequent AS event (∼40%), but many IRs have relatively low read coverage and are less well-represented in assembled transcripts. Additionally, ∼51% of Arabidopsis genes produce AS transcripts which do not involve IR. Therefore, the significance of IR in generating transcript diversity was generally overestimated in previous assessments. IR analysis allowed the identification of a large set of cryptic introns inside annotated coding exons. Importantly, a significant fraction of these cryptic introns are spliced out in frame, indicating a role in protein diversity. Furthermore, we show extensive AS coupled to nonsense-mediated decay in AFC2, encoding a highly conserved LAMMER kinase which phosphorylates splicing factors, thus establishing a complex loop in AS regulation. We provide the most comprehensive analysis of AS to date which will serve as a valuable resource for the plant community to study transcriptome complexity and gene regulation.
Figures
References
-
- Ali GS, Reddy AS 2008. Regulation of alternative splicing of pre-mRNAs by stresses. Curr Top Microbiol Immunol 326: 257–275 - PubMed
-
- The Arabidopsis Genome Initiative 2000. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408: 796–815 - PubMed
-
- Barbazuk WB, Fu Y, McGinnis KM 2008. Genome-wide analyses of alternative splicing in plants: Opportunities and challenges. Genome Res 18: 1381–1392 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials