

Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8
- PMID: 22391568
- PMCID: PMC3371110
- DOI: 10.1038/onc.2012.81
Histone deacetylase inhibitors suppress mutant p53 transcription via histone deacetylase 8
Abstract
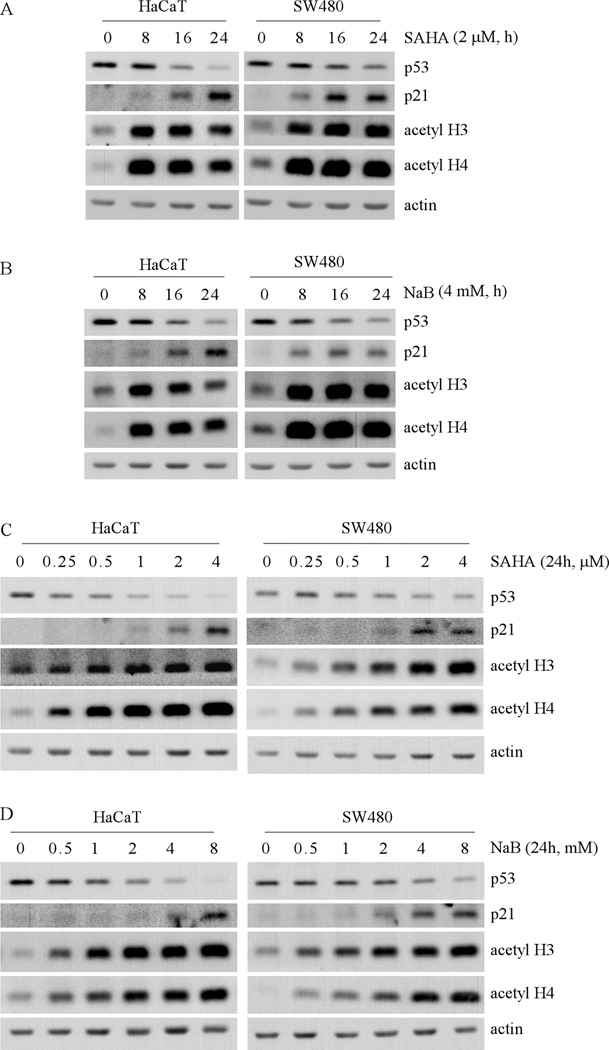
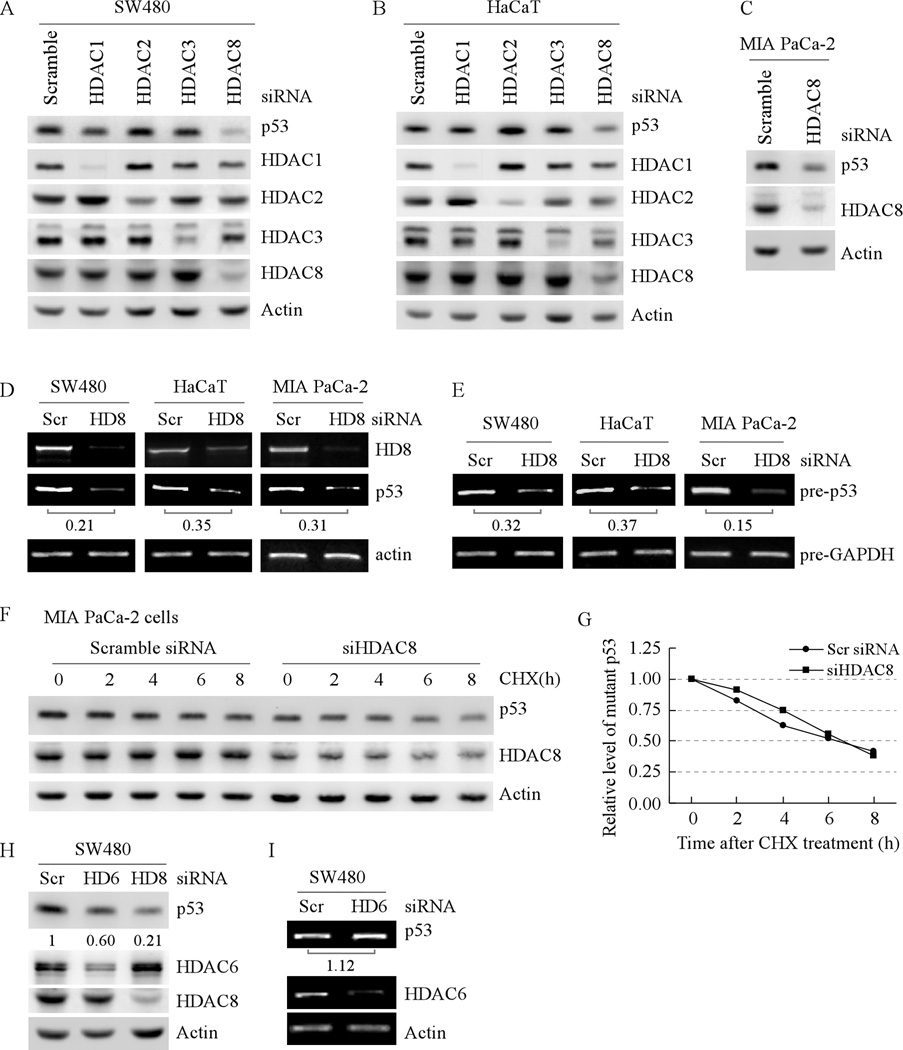
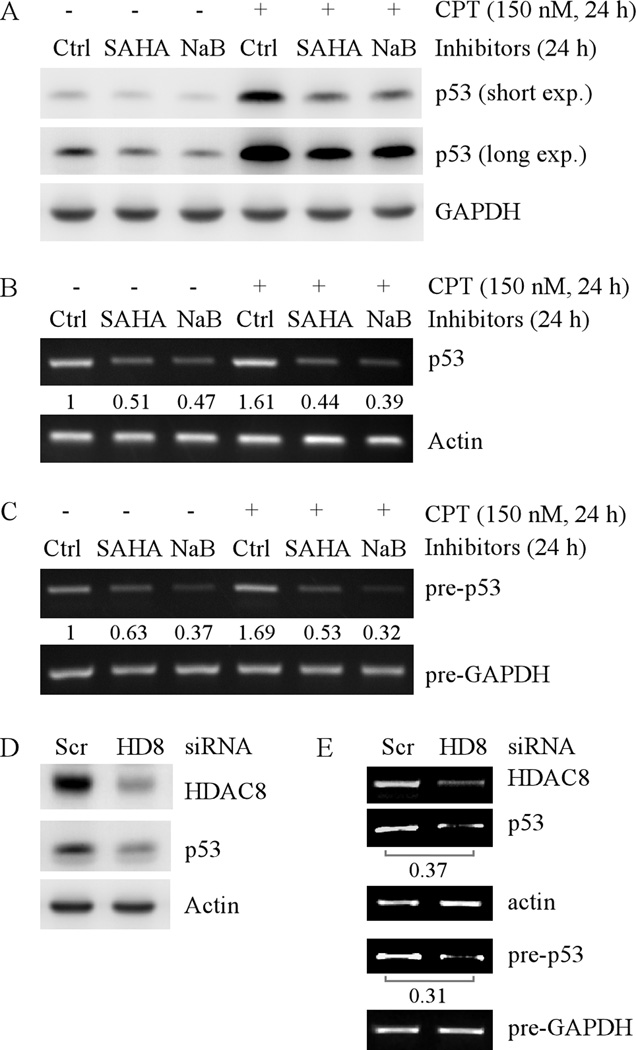
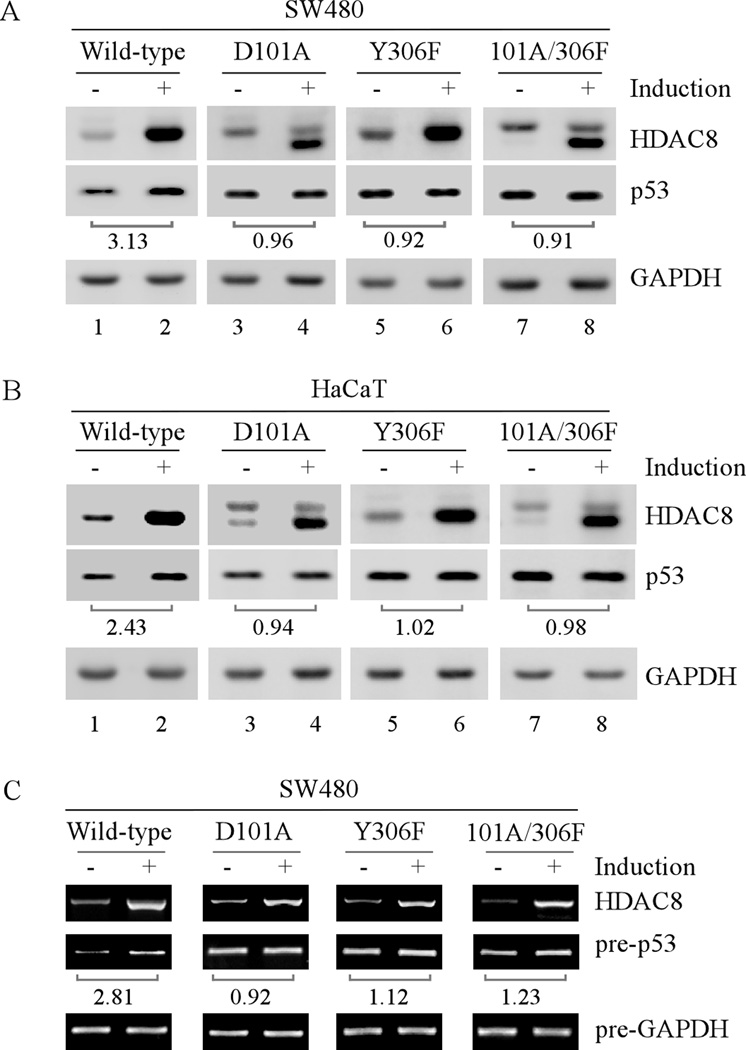
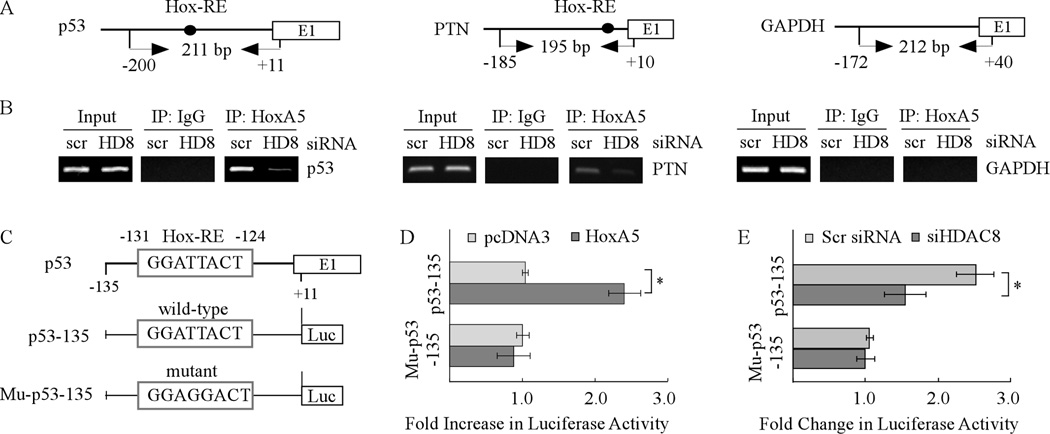
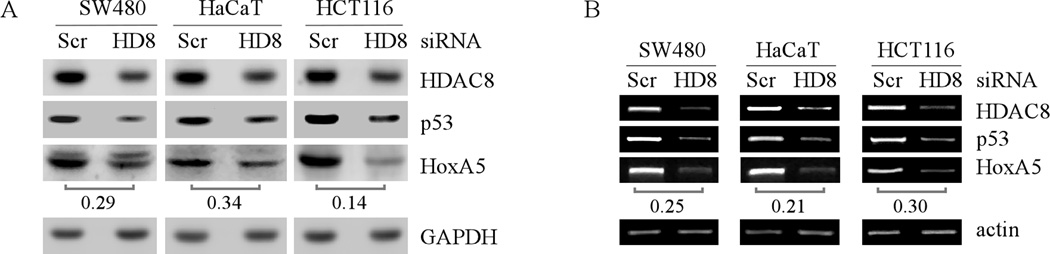
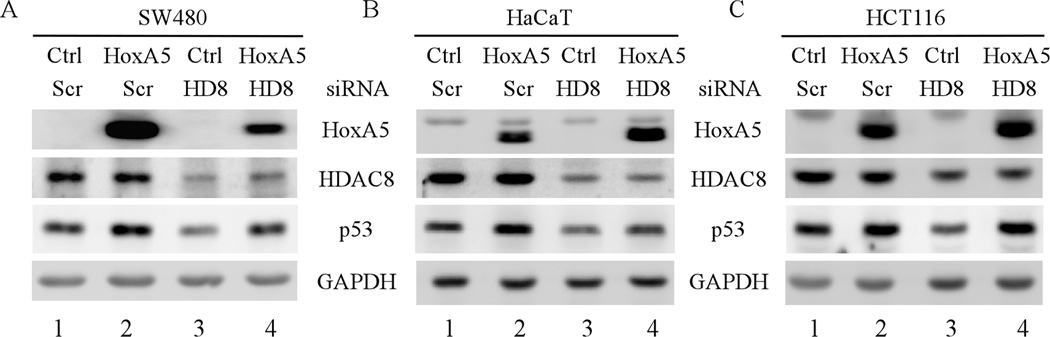
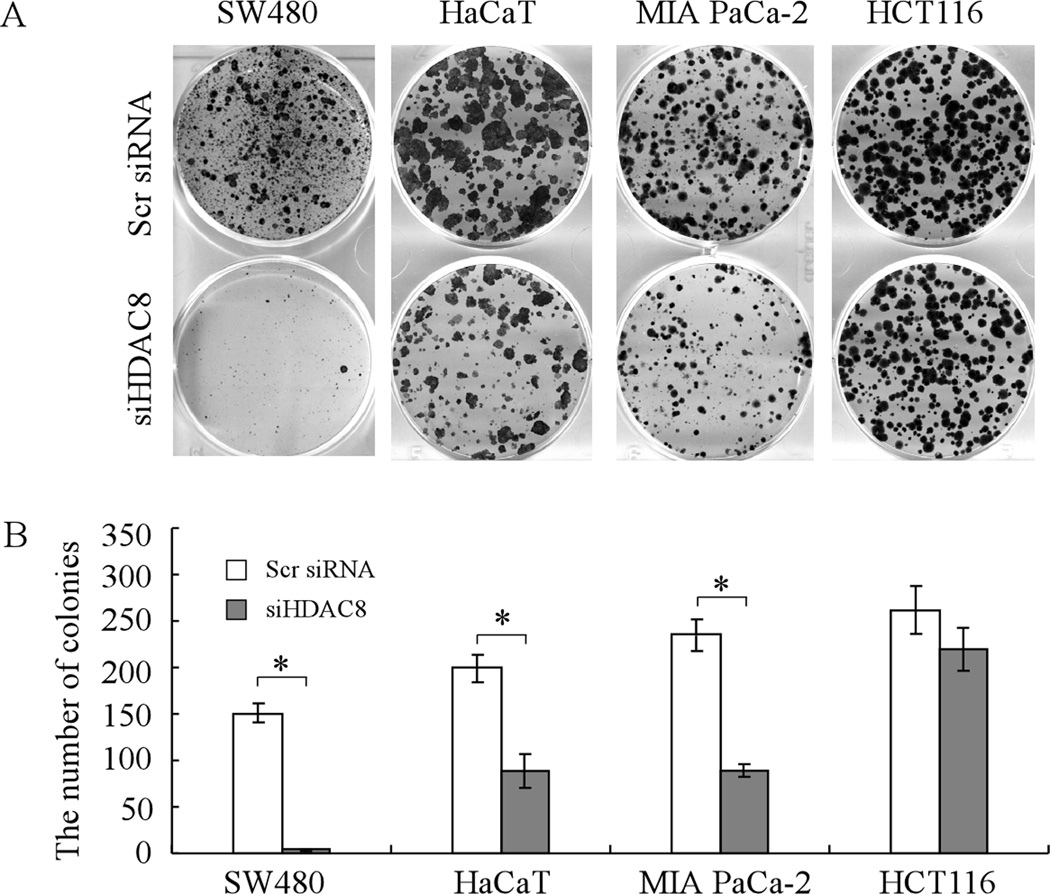
Mutation of the p53 gene is the most common genetic alteration in human cancer and contributes to malignant process by enhancing transformed properties of cells and resistance to anticancer therapy. Mutant p53 is often highly expressed in tumor cells at least, in part, due to its increased half-life. However, whether mutant p53 expression is regulated by other mechanisms in tumors is unclear. Here we found that histone deacetylase (HDAC) inhibitors suppress both wild-type and mutant p53 transcription in time- and dose-dependent manners. Consistent with this, the levels of wild-type and mutant p53 proteins are decreased upon treatment with HDAC inhibitors. Importantly, we found that upon knockdown of each class I HDAC, only HDAC8 knockdown leads to decreased expression of wild-type and mutant p53 proteins and transcripts. Conversely, we found that ectopic expression of wild-type, but not mutant HDAC8, leads to increased transcription of p53. Furthermore, we found that knockdown of HDAC8 results in reduced expression of HoxA5 and consequently, attenuated ability of HoxA5 to activate p53 transcription, which can be rescued by ectopic expression of HoxA5. Because of the fact that HDAC8 is required for expression of both wild-type and mutant p53, we found that targeted disruption of HDAC8 expression remarkably triggers proliferative defect in cells with a mutant, but not wild-type, p53. Together, our data uncover a regulatory mechanism of mutant p53 transcription via HDAC8 and suggest that HDAC inhibitors and especially HDAC8-targeting agents might be explored as an adjuvant for tumors carrying a mutant p53.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. - PubMed
-
- Halkidou K, Gaughan L, Cook S, Leung HY, Neal DE, Robson CN. Upregulation and nuclear recruitment of HDAC1 in hormone refractory prostate cancer. Prostate. 2004;59:177–189. - PubMed
-
- Song J, Noh JH, Lee JH, Eun JW, Ahn YM, Kim SY, et al. Increased expression of histone deacetylase 2 is found in human gastric cancer. APMIS. 2005;113:264–268. - PubMed
-
- Huang BH, Laban M, Leung CH, Lee L, Lee CK, Salto-Tellez M, et al. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005;12:395–404. - PubMed
-
- Hrzenjak A, Moinfar F, Kremser ML, Strohmeier B, Staber PB, Zatloukal K, et al. Valproate inhibition of histone deacetylase 2 affects differentiation and decreases proliferation of endometrial stromal sarcoma cells. Mol Cancer Ther. 2006;5:2203–2210. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
