

Infantile cerebellar-retinal degeneration associated with a mutation in mitochondrial aconitase, ACO2
- PMID: 22405087
- PMCID: PMC3309186
- DOI: 10.1016/j.ajhg.2012.01.009
Infantile cerebellar-retinal degeneration associated with a mutation in mitochondrial aconitase, ACO2
Abstract
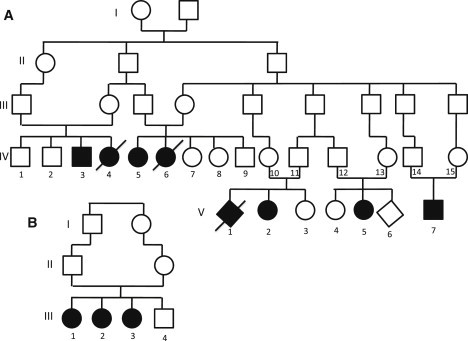
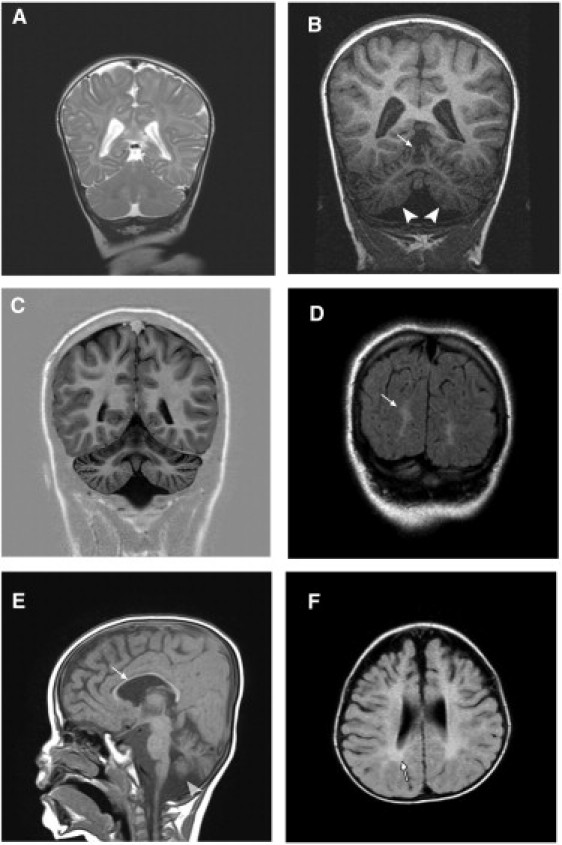
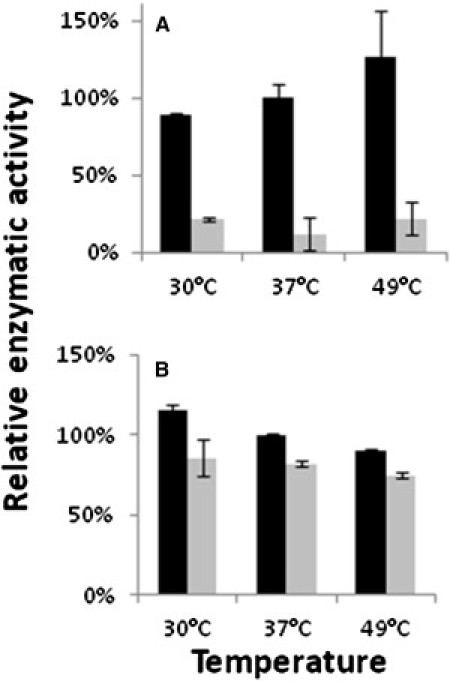
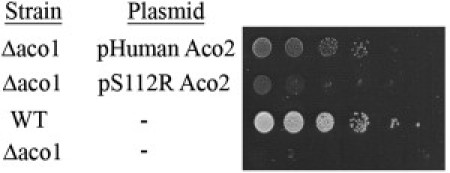
Degeneration of the cerebrum, cerebellum, and retina in infancy is part of the clinical spectrum of lysosomal storage disorders, mitochondrial respiratory chain defects, carbohydrate glycosylation defects, and infantile neuroaxonal dystrophy. We studied eight individuals from two unrelated families who presented at 2-6 months of age with truncal hypotonia and athetosis, seizure disorder, and ophthalmologic abnormalities. Their course was characterized by failure to acquire developmental milestones and culminated in profound psychomotor retardation and progressive visual loss, including optic nerve and retinal atrophy. Despite their debilitating state, the disease was compatible with survival of up to 18 years. Laboratory investigations were normal, but the oxidation of glutamate by muscle mitochondria was slightly reduced. Serial brain MRI displayed progressive, prominent cerebellar atrophy accompanied by thinning of the corpus callosum, dysmyelination, and frontal and temporal cortical atrophy. Homozygosity mapping followed by whole-exome sequencing disclosed a Ser112Arg mutation in ACO2, encoding mitochondrial aconitase, a component of the Krebs cycle. Specific aconitase activity in the individuals' lymphoblasts was severely reduced. Under restrictive conditions, the mutant human ACO2 failed to complement a yeast ACO1 deletion strain, whereas the wild-type human ACO2 succeeded, indicating that this mutation is pathogenic. Thus, a defect in mitochondrial aconitase is associated with an infantile neurodegenerative disorder affecting mainly the cerebellum and retina. In the absence of noninvasive biomarkers, determination of the ACO2 sequence or of aconitase activity in lymphoblasts are warranted in similarly affected individuals, based on clinical and neuroradiologic grounds.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Nardocci N., Verga M.L., Binelli S., Zorzi G., Angelini L., Bugiani O. Neuronal ceroid-lipofuscinosis: a clinical and morphological study of 19 patients. Am. J. Med. Genet. 1995;57:137–141. - PubMed
-
- Uziel G., Moroni I., Lamantea E., Fratta G.M., Ciceri E., Carrara F., Zeviani M. Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: a clinical, biochemical, and molecular study in six families. J. Neurol. Neurosurg. Psychiatry. 1997;63:16–22. - PMC - PubMed
-
- Jaeken J., Matthijs G. Congenital disorders of glycosylation: a rapidly expanding disease family. Annu. Rev. Genomics Hum. Genet. 2007;8:261–278. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
