

G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer
- PMID: 22406531
- PMCID: PMC3314447
- DOI: 10.1172/JCI57349
G9a interacts with Snail and is critical for Snail-mediated E-cadherin repression in human breast cancer
Abstract
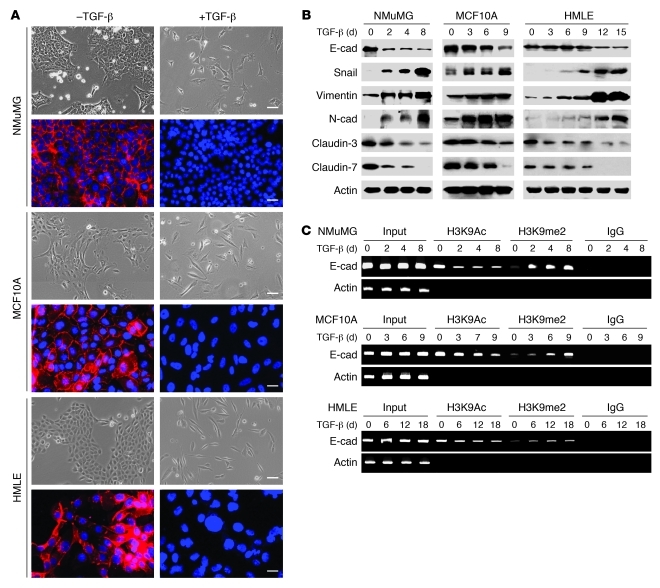
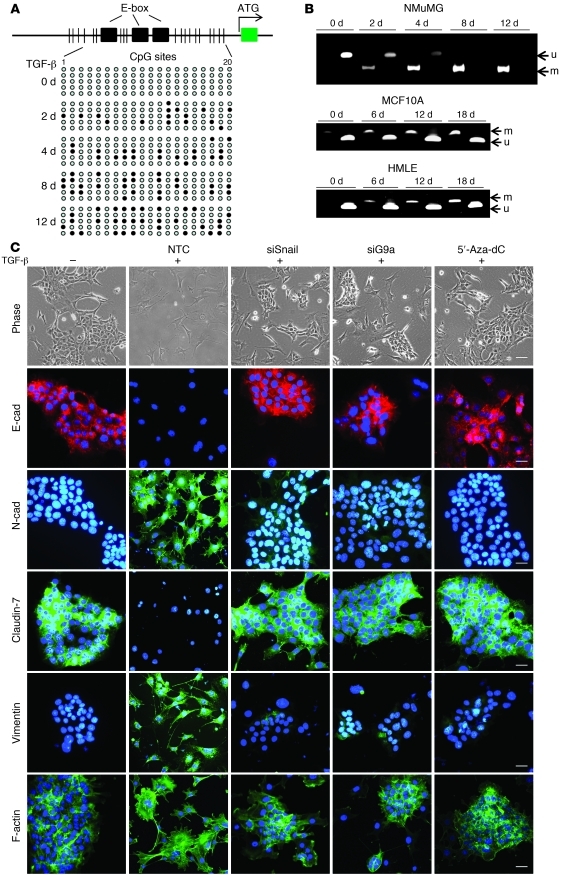
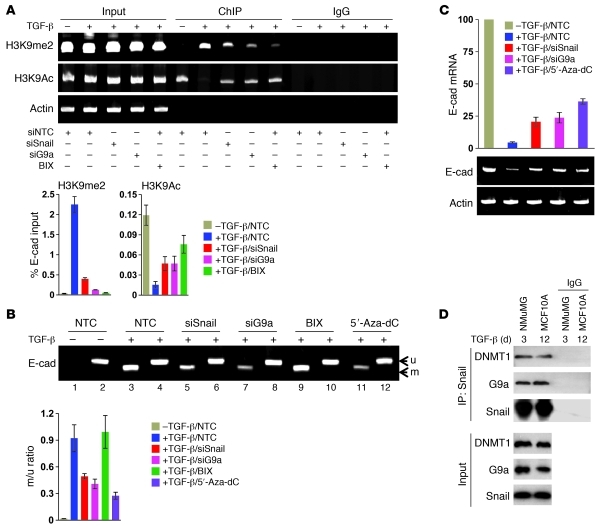
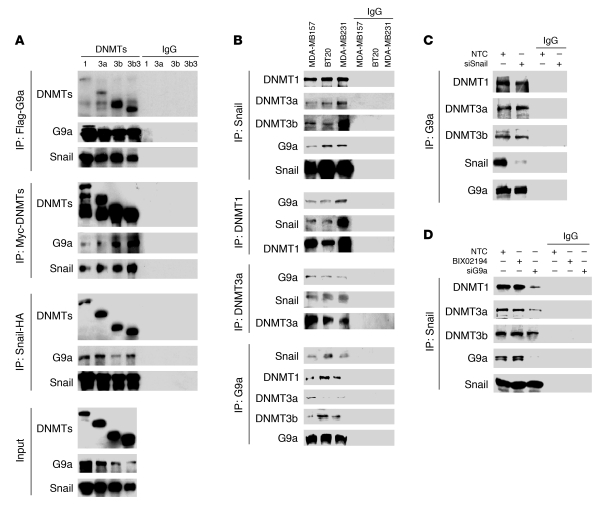
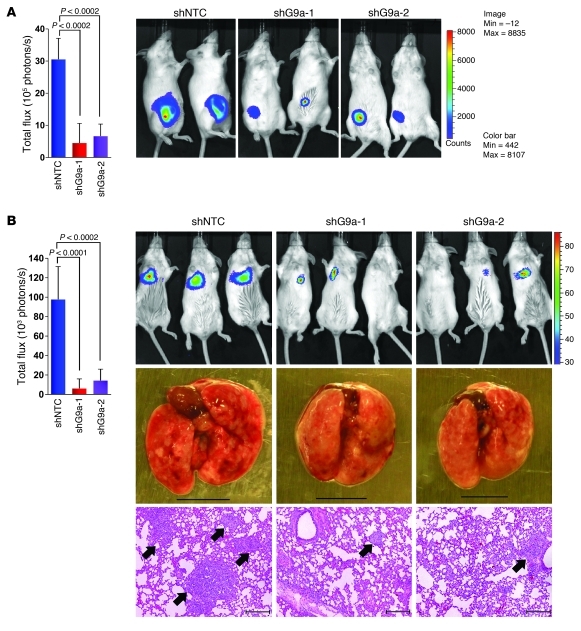
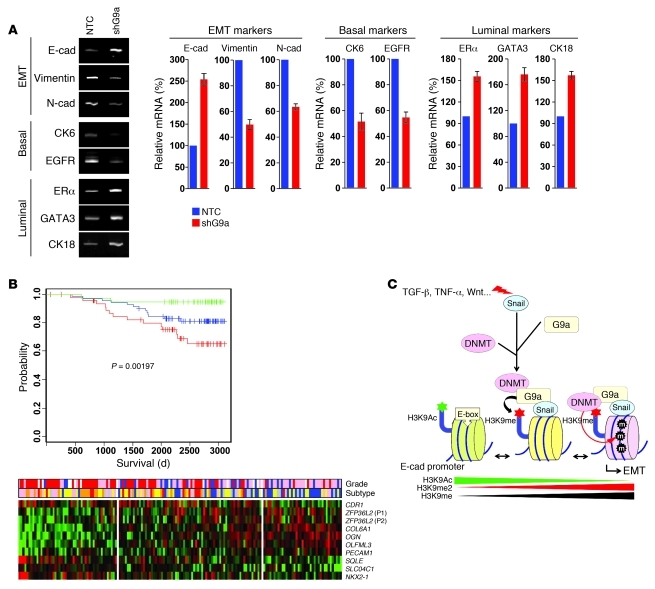
Breast cancers are highly heterogeneous but can be grouped into subtypes based on several criteria, including level of expression of certain markers. Claudin-low breast cancer (CLBC) is associated with early metastasis and resistance to chemotherapy, while gene profiling indicates it is characterized by the expression of markers of epithelial-mesenchymal transition (EMT) - a phenotypic conversion linked with metastasis. Although the epigenetic program controlling the phenotypic and cellular plasticity of EMT remains unclear, one contributor may be methylation of the E-cadherin promoter, resulting in decreased E-cadherin expression, a hallmark of EMT. Indeed, reduced E-cadherin often occurs in CLBC and may contribute to the early metastasis and poor patient survival associated with this disease. Here, we have determined that methylation of histone H3 on lysine 9 (H3K9me2) is critical for promoter DNA methylation of E-cadherin in three TGF-β-induced EMT model cell lines, as well as in CLBC cell lines. Further, Snail interacted with G9a, a major euchromatin methyltransferase responsible for H3K9me2, and recruited G9a and DNA methyltransferases to the E-cadherin promoter for DNA methylation. Knockdown of G9a restored E-cadherin expression by suppressing H3K9me2 and blocking DNA methylation. This resulted in inhibition of cell migration and invasion in vitro and suppression of tumor growth and lung colonization in in vivo models of CLBC metastasis. Our study not only reveals a critical mechanism underlying the epigenetic regulation of EMT but also paves a way for the development of new treatment strategies for CLBC.
Figures
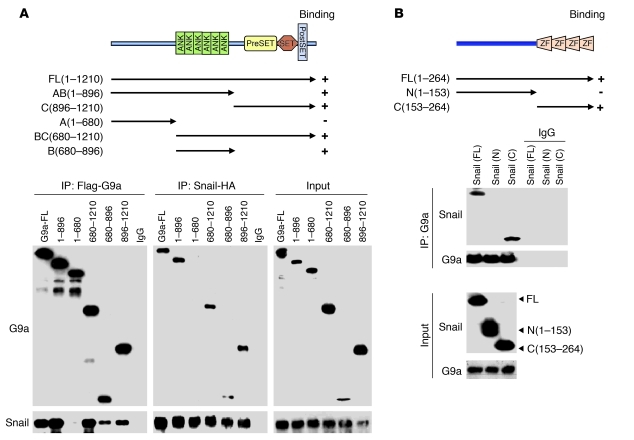
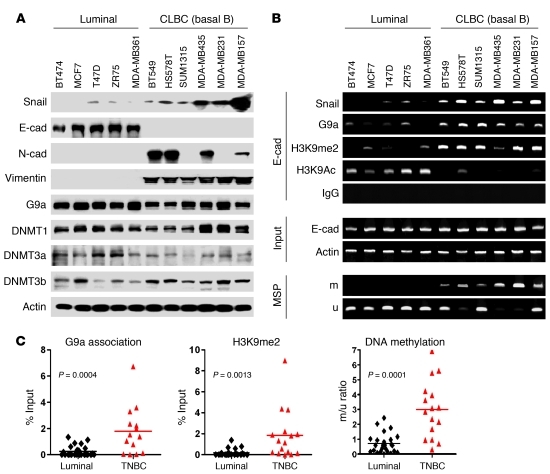
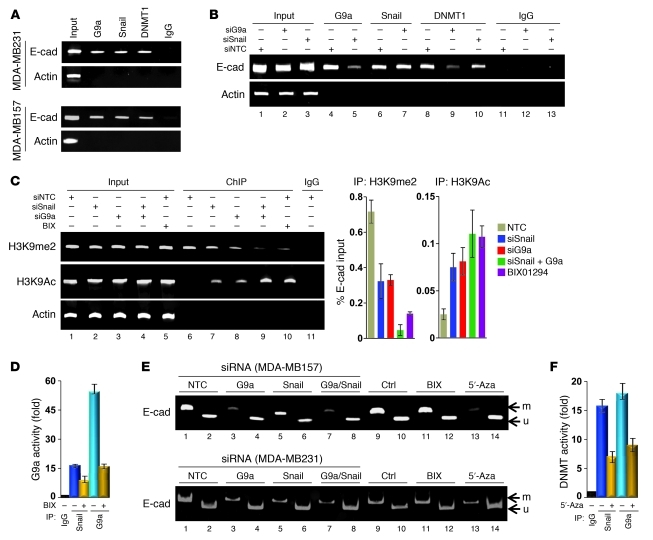
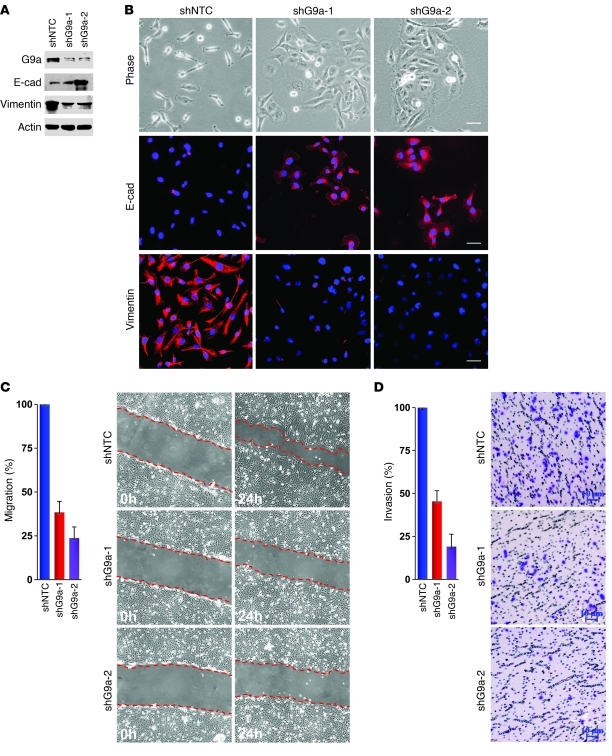
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
