

An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice
- PMID: 22406536
- PMCID: PMC3314459
- DOI: 10.1172/JCI59743
An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice
Abstract
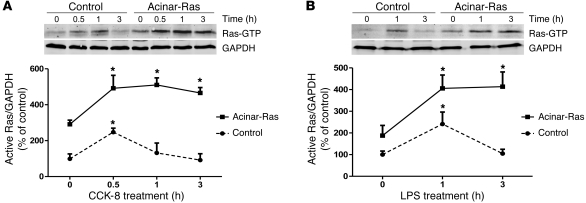
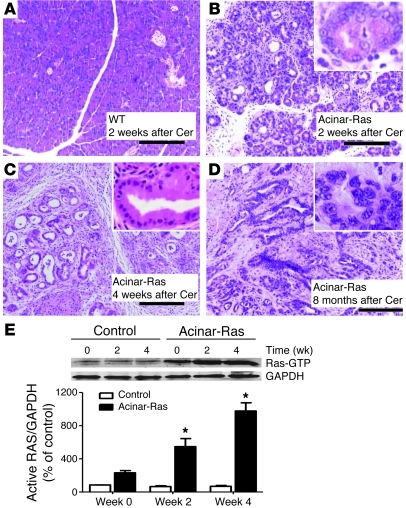
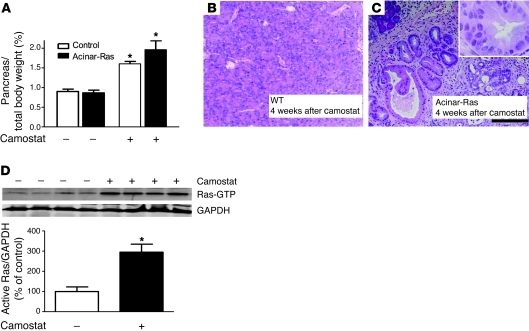
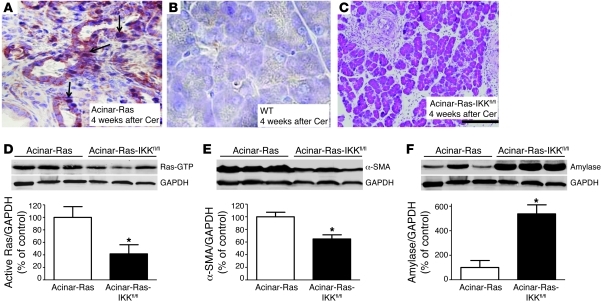
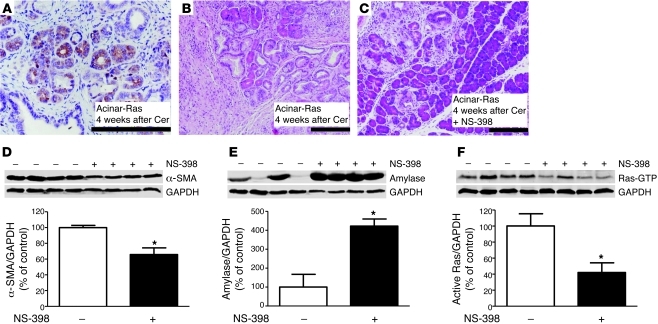
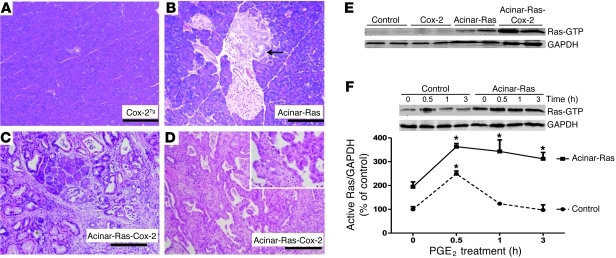
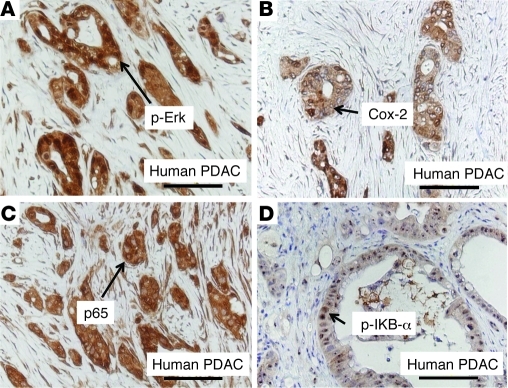
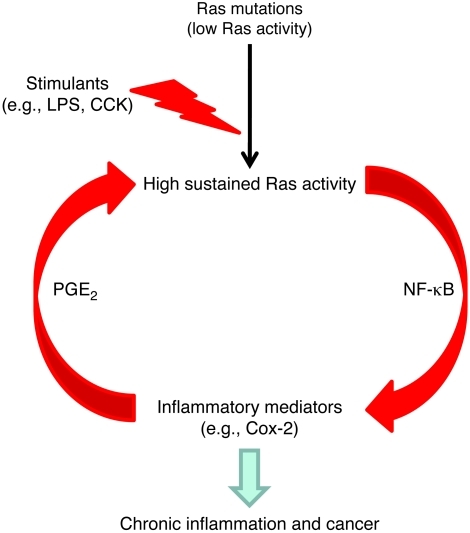
Genetic mutations that give rise to active mutant forms of Ras are oncogenic and found in several types of tumor. However, such mutations are not clear biomarkers for disease, since they are frequently detected in healthy individuals. Instead, it has become clear that elevated levels of Ras activity are critical for Ras-induced tumorigenesis. However, the mechanisms underlying the production of pathological levels of Ras activity are unclear. Here, we show that in the presence of oncogenic Ras, inflammatory stimuli initiate a positive feedback loop involving NF-κB that further amplifies Ras activity to pathological levels. Stimulation of Ras signaling by typical inflammatory stimuli was transient and had no long-term sequelae in wild-type mice. In contrast, these stimuli generated prolonged Ras signaling and led to chronic inflammation and precancerous pancreatic lesions (PanINs) in mice expressing physiological levels of oncogenic K-Ras. These effects of inflammatory stimuli were disrupted by deletion of inhibitor of NF-κB kinase 2 (IKK2) or inhibition of Cox-2. Likewise, expression of active IKK2 or Cox-2 or treatment with LPS generated chronic inflammation and PanINs only in mice expressing oncogenic K-Ras. The data support the hypothesis that in the presence of oncogenic Ras, inflammatory stimuli trigger an NF-κB-mediated positive feedback mechanism involving Cox-2 that amplifies Ras activity to pathological levels. Because a large proportion of the adult human population possesses Ras mutations in tissues including colon, pancreas, and lung, disruption of this positive feedback loop may be an important strategy for cancer prevention.
Figures

References
-
- Lu X, Xu T, Qian J, Wen X, Wu D. Detecting K-ras and p53 gene mutation from stool and pancreatic juice for diagnosis of early pancreatic cancer. Chin Med J (Engl). 2002;115(11):1632–1636. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
