

Genome-wide analysis of long noncoding RNA stability
- PMID: 22406755
- PMCID: PMC3337434
- DOI: 10.1101/gr.131037.111
Genome-wide analysis of long noncoding RNA stability
Abstract
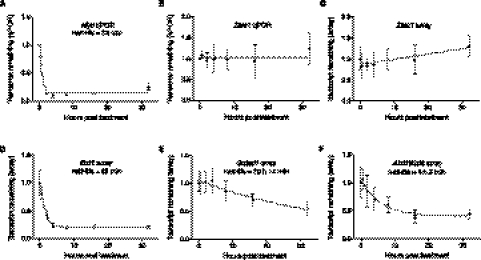
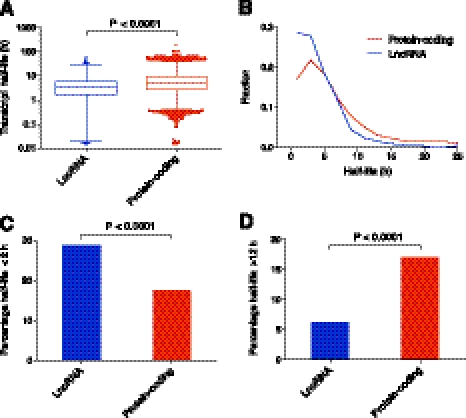
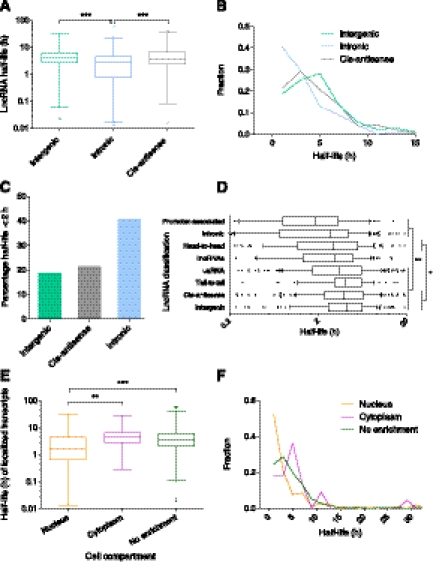
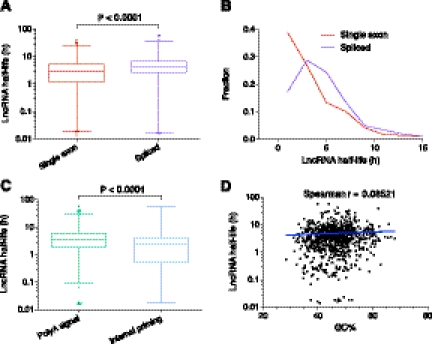
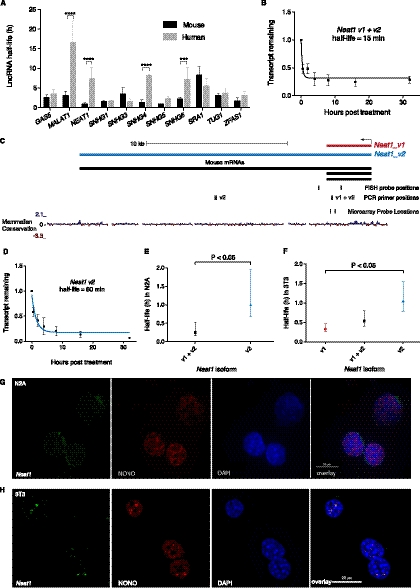
Transcriptomic analyses have identified tens of thousands of intergenic, intronic, and cis-antisense long noncoding RNAs (lncRNAs) that are expressed from mammalian genomes. Despite progress in functional characterization, little is known about the post-transcriptional regulation of lncRNAs and their half-lives. Although many are easily detectable by a variety of techniques, it has been assumed that lncRNAs are generally unstable, but this has not been examined genome-wide. Utilizing a custom noncoding RNA array, we determined the half-lives of ∼800 lncRNAs and ∼12,000 mRNAs in the mouse Neuro-2a cell line. We find only a minority of lncRNAs are unstable. LncRNA half-lives vary over a wide range, comparable to, although on average less than, that of mRNAs, suggestive of complex metabolism and widespread functionality. Combining half-lives with comprehensive lncRNA annotations identified hundreds of unstable (half-life < 2 h) intergenic, cis-antisense, and intronic lncRNAs, as well as lncRNAs showing extreme stability (half-life > 16 h). Analysis of lncRNA features revealed that intergenic and cis-antisense RNAs are more stable than those derived from introns, as are spliced lncRNAs compared to unspliced (single exon) transcripts. Subcellular localization of lncRNAs indicated widespread trafficking to different cellular locations, with nuclear-localized lncRNAs more likely to be unstable. Surprisingly, one of the least stable lncRNAs is the well-characterized paraspeckle RNA Neat1, suggesting Neat1 instability contributes to the dynamic nature of this subnuclear domain. We have created an online interactive resource (http://stability.matticklab.com) that allows easy navigation of lncRNA and mRNA stability profiles and provides a comprehensive annotation of ~7200 mouse lncRNAs.
Figures
References
-
- Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI 2002. Directed proteomic analysis of the human nucleolus. Curr Biol 12: 1–11 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources