

Microcephaly, intellectual impairment, bilateral vesicoureteral reflux, distichiasis, and glomuvenous malformations associated with a 16q24.3 contiguous gene deletion and a Glomulin mutation
- PMID: 22407726
- PMCID: PMC3314153
- DOI: 10.1002/ajmg.a.35229
Microcephaly, intellectual impairment, bilateral vesicoureteral reflux, distichiasis, and glomuvenous malformations associated with a 16q24.3 contiguous gene deletion and a Glomulin mutation
Abstract
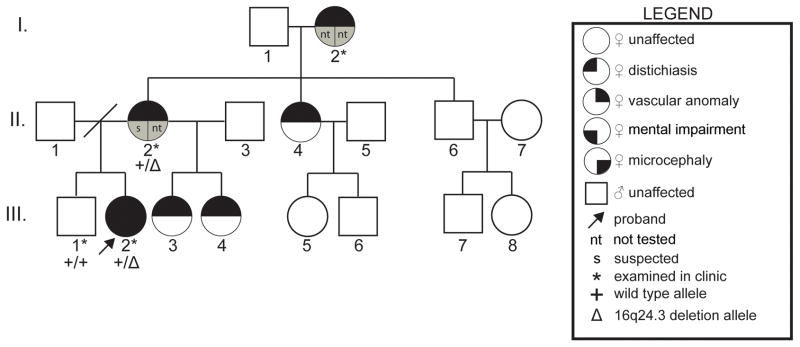
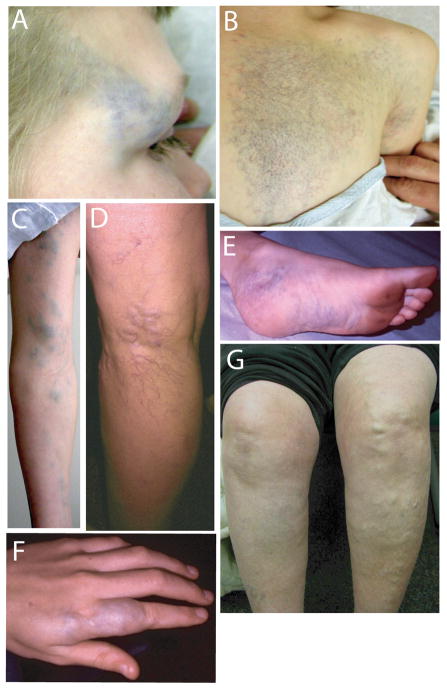
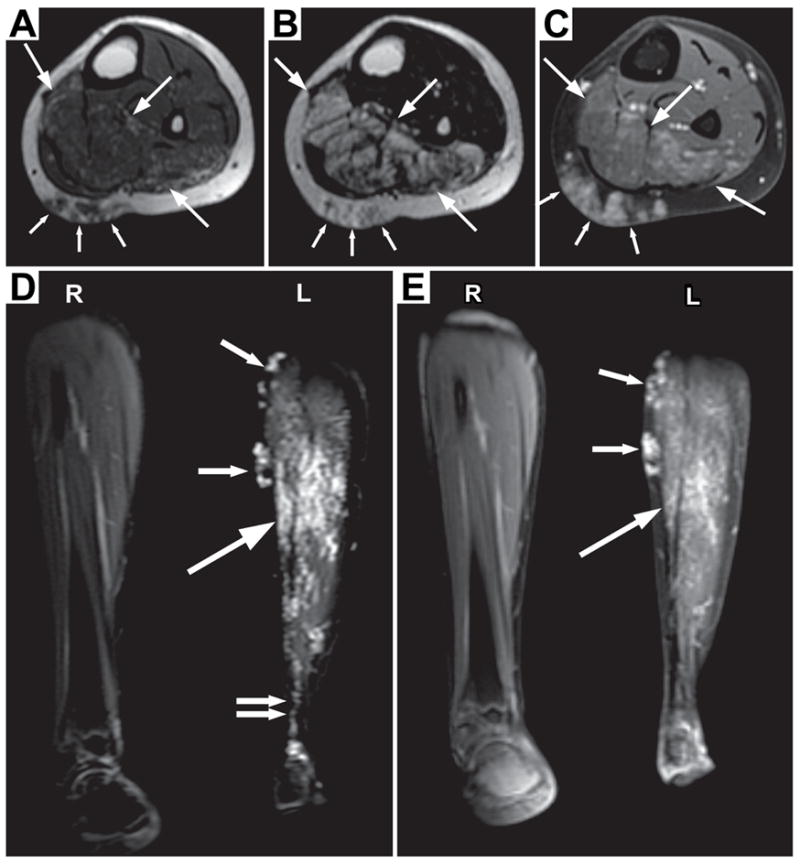
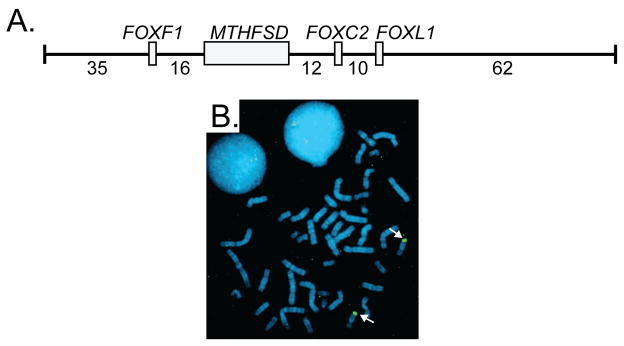
Two hereditary syndromes, lymphedema-distichiasis (LD) syndrome and blepharo-chelio-dontic (BCD) syndrome include the aberrant growth of eyelashes from the meibomian glands, known as distichiasis. LD is an autosomal dominant syndrome primarily characterized by distichiasis and the onset of lymphedema usually during puberty. Mutations in the forkhead transcription factor FOXC2 are the only known cause of LD. BCD syndrome consists of autosomal dominant abnormalities of the eyelid, lip, and teeth, and the etiology remains unknown. In this report, we describe a proband that presented with distichiasis, microcephaly, bilateral grade IV vesicoureteral reflux requiring ureteral re-implantation, mild intellectual impairment and apparent glomuvenous malformations (GVM). Distichiasis was present in three generations of the proband's maternal side of the family. The GVMs were severe in the proband, and maternal family members exhibited lower extremity varicosities of variable degree. A GLMN (glomulin) gene mutation was identified in the proband that accounts for the observed GVMs; no other family member could be tested. TIE2 sequencing revealed no mutations. In the proband, an additional submicroscopic 265 kb contiguous gene deletion was identified in 16q24.3, located 609 kb distal to the FOXC2 locus, which was inherited from the proband's mother. The deletion includes the C16ORF95, FBXO31, MAP1LC3B, and ZCCHC14 loci and 115 kb of a gene desert distal to FOXC2 and FOXL1. Thus, it is likely that the microcephaly, distichiasis, vesicoureteral, and intellectual impairment in this family may be caused by the deletion of one or more of these genes and/or deletion of distant cis-regulatory elements of FOXC2 expression.
Copyright © 2012 Wiley Periodicals, Inc.
Figures
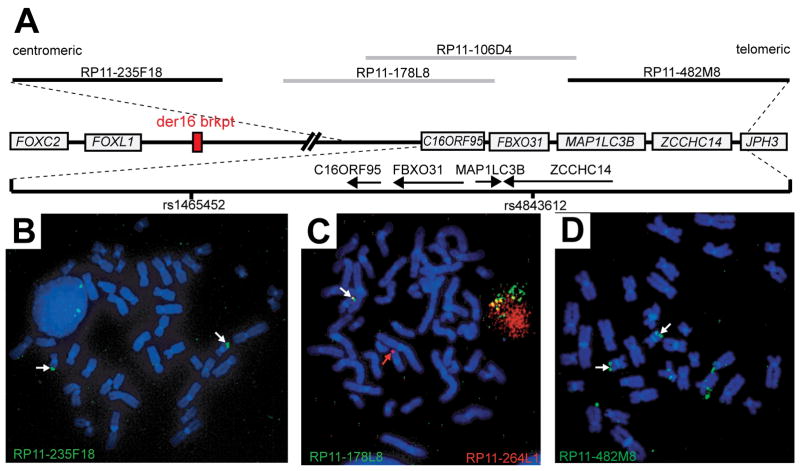
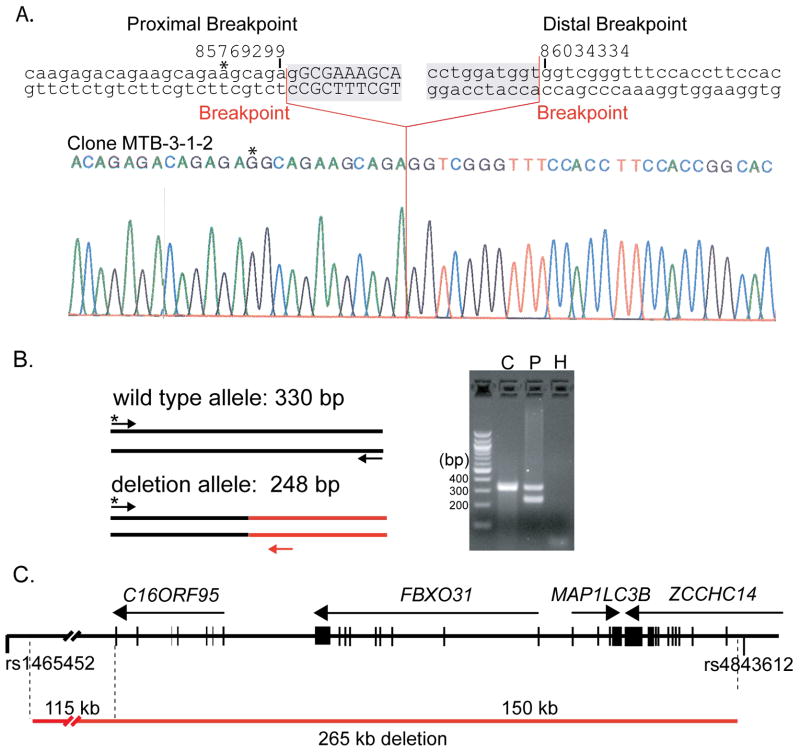
References
-
- Bell R, Brice G, Child AH, Murday VA, Mansour S, Sandy CJ, Collin JR, Brady AF, Callen DF, Burnand K, Mortimer P, Jeffery S. Analysis of lymphoedema-distichiasis families for FOXC2 mutations reveals small insertions and deletions throughout the gene. Hum Genet. 2001;108:546–551. - PubMed
-
- Boon LM, Mulliken JB, Enjolras O, Vikkula M. Glomuvenous malformation (glomangioma) and venous malformation: distinct clinicopathologic and genetic entities. Arch Dermatol. 2004;140:971–976. - PubMed
-
- Brooks BP, Dagenais SL, Nelson CC, Glynn MW, Caulder MS, Downs CA, Glover TW. Mutation of the FOXC2 gene in familial distichiasis. J Aapos. 2003;7:354–357. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
