

Developmental disorders of the midbrain and hindbrain
- PMID: 22408608
- PMCID: PMC3294267
- DOI: 10.3389/fnana.2012.00007
Developmental disorders of the midbrain and hindbrain
Abstract
Malformations of the midbrain (MB) and hindbrain (HB) have become topics of considerable interest in the neurology and neuroscience literature in recent years. The combined advances of imaging and molecular biology have improved analyses of structures in these areas of the central nervous system, while advances in genetics have made it clear that malformations of these structures are often associated with dysfunction or malformation of other organ systems. This review focuses upon the importance of communication between clinical researchers and basic scientists in the advancement of knowledge of this group of disorders. Disorders of anteroposterior (AP) patterning, cerebellar hypoplasias, disorders associated with defects of the pial limiting membrane (cobblestone cortex), disorders of the Reelin pathway, and disorders of the primary cilium/basal body organelle (molar tooth malformations) are the main focus of the review.
Keywords: cerebellum; hindbrain; malformations; midbrain.
Figures


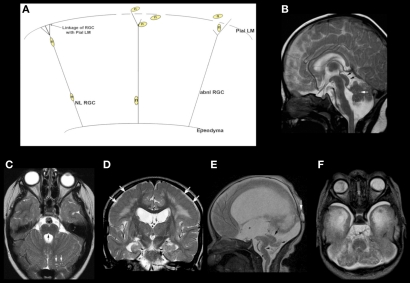
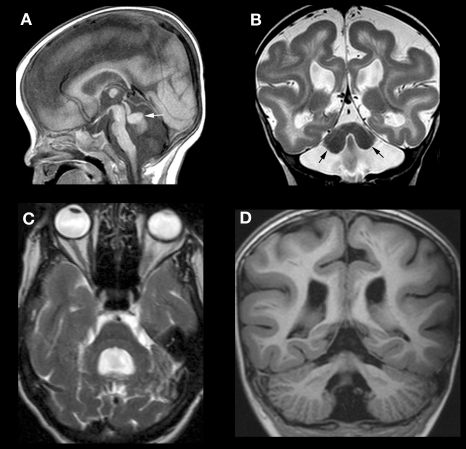
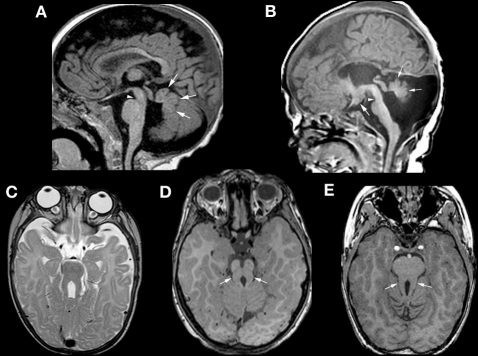
References
-
- Aldinger K. A., Lehmann O. J., Hudgins L., Chizhikov V. V., Bassuk A. G., Ades L. C., Krantz I. D., Dobyns W. B., Millen K. J. (2009). FOXC1 is required for normal cerebellar development and is a major contributor to chromosome 6p25.3 Dandy-Walker malformation. Nat. Genet. 41, 1037–1042 10.1038/ng.422 - DOI - PMC - PubMed
-
- Aranda-Orgillés B., Trockenbacher A., Winter J., Aigner J., Köhler A., Jastrzebska E., Stahl J., Müller E.-C., Otto A., Wanker E., Schneider R., Schweiger S. (2008). The Opitz syndrome gene product MID1 assembles a microtubule-associated ribonucleoprotein complex. Hum. Genet. 123, 163–176 10.1007/s00439-007-0456-6 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous