

Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors
- PMID: 22411871
- PMCID: PMC3359741
- DOI: 10.1182/blood-2011-10-385658
Rac2-MRC-cIII-generated ROS cause genomic instability in chronic myeloid leukemia stem cells and primitive progenitors
Abstract
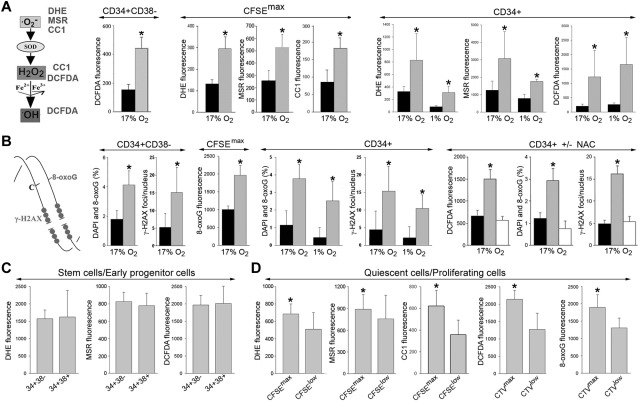
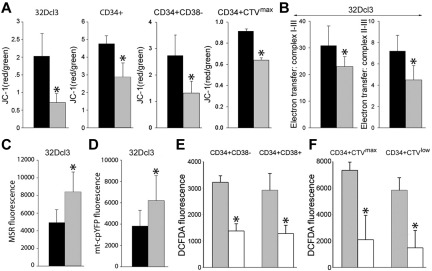
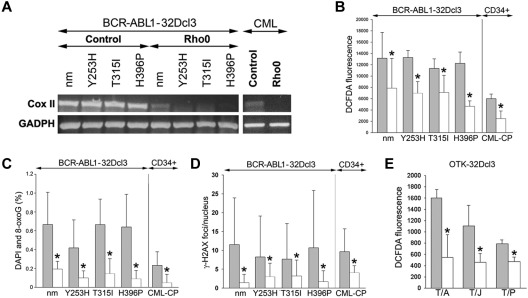
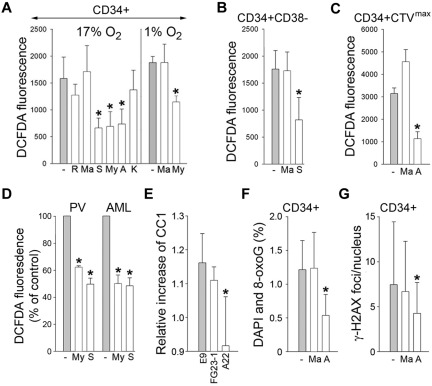
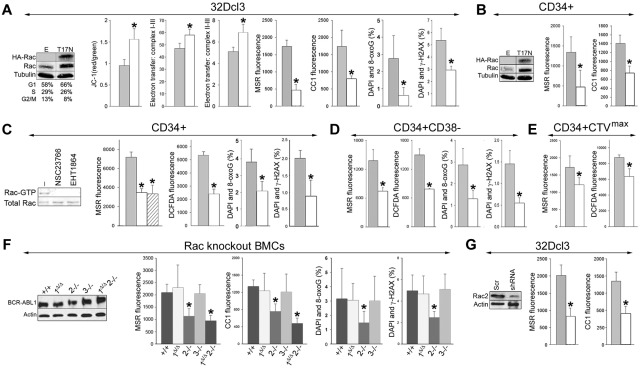
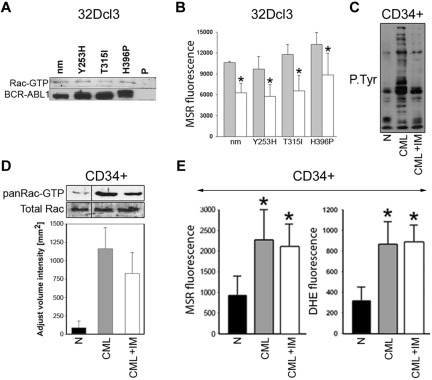
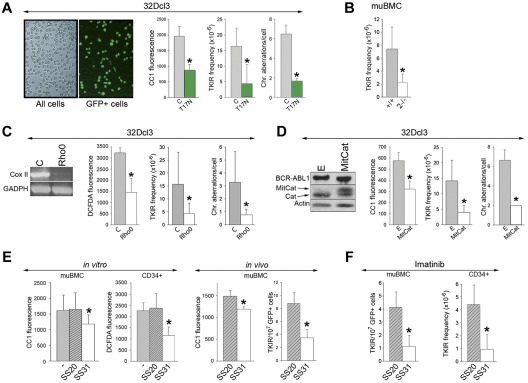
Chronic myeloid leukemia in chronic phase (CML-CP) is induced by BCR-ABL1 oncogenic tyrosine kinase. Tyrosine kinase inhibitors eliminate the bulk of CML-CP cells, but fail to eradicate leukemia stem cells (LSCs) and leukemia progenitor cells (LPCs) displaying innate and acquired resistance, respectively. These cells may accumulate genomic instability, leading to disease relapse and/or malignant progression to a fatal blast phase. In the present study, we show that Rac2 GTPase alters mitochondrial membrane potential and electron flow through the mitochondrial respiratory chain complex III (MRC-cIII), thereby generating high levels of reactive oxygen species (ROS) in CML-CP LSCs and primitive LPCs. MRC-cIII-generated ROS promote oxidative DNA damage to trigger genomic instability, resulting in an accumulation of chromosomal aberrations and tyrosine kinase inhibitor-resistant BCR-ABL1 mutants. JAK2(V617F) and FLT3(ITD)-positive polycythemia vera cells and acute myeloid leukemia cells also produce ROS via MRC-cIII. In the present study, inhibition of Rac2 by genetic deletion or a small-molecule inhibitor and down-regulation of mitochondrial ROS by disruption of MRC-cIII, expression of mitochondria-targeted catalase, or addition of ROS-scavenging mitochondria-targeted peptide aptamer reduced genomic instability. We postulate that the Rac2-MRC-cIII pathway triggers ROS-mediated genomic instability in LSCs and primitive LPCs, which could be targeted to prevent the relapse and malignant progression of CML.
Figures
References
-
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. - PubMed
-
- Melo JV, Barnes DJ. Chronic myeloid leukaemia as a model of disease evolution in human cancer. Nat Rev Cancer. 2007;7(6):441–453. - PubMed
-
- Slupphaug G, Kavli B, Krokan HE. The interacting pathways for prevention and repair of oxidative DNA damage. Mutat Res. 2003;531(1-2):231–251. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- CA123014/CA/NCI NIH HHS/United States
- CA134458/CA/NCI NIH HHS/United States
- R21 CA133646/CA/NCI NIH HHS/United States
- T32 HL007574/HL/NHLBI NIH HHS/United States
- T32 HL07574-29/HL/NHLBI NIH HHS/United States
- R01 CA123014/CA/NCI NIH HHS/United States
- R01 DK062757/DK/NIDDK NIH HHS/United States
- CA134660/CA/NCI NIH HHS/United States
- CA133646/CA/NCI NIH HHS/United States
- C1174/A11008/CRUK_/Cancer Research UK/United Kingdom
- DK62757/DK/NIDDK NIH HHS/United States
- CSO_/Chief Scientist Office/United Kingdom
- R01 CA134660/CA/NCI NIH HHS/United States
- 11008/CRUK_/Cancer Research UK/United Kingdom
- R01 CA134458/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
