

Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae
- PMID: 22419079
- PMCID: PMC3296254
- DOI: 10.1534/genetics.111.133306
Regulation of amino acid, nucleotide, and phosphate metabolism in Saccharomyces cerevisiae
Abstract
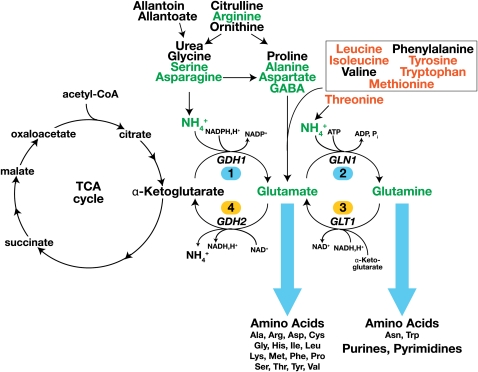
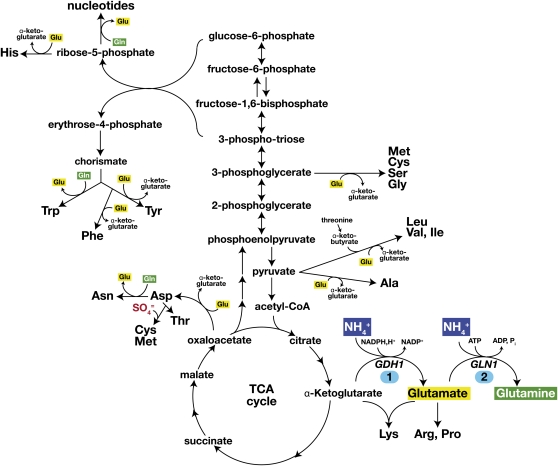
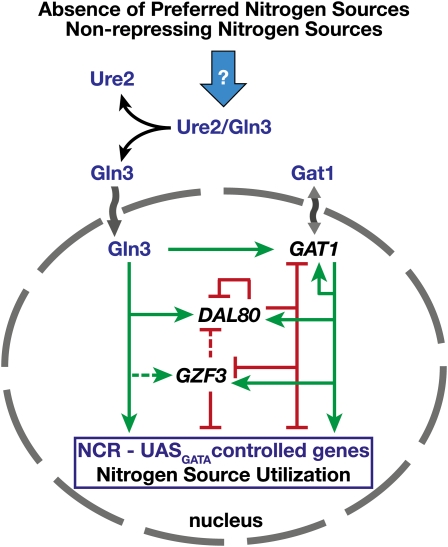
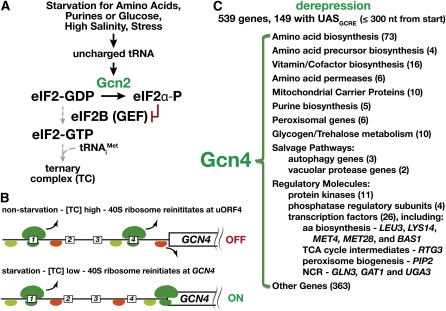
Ever since the beginning of biochemical analysis, yeast has been a pioneering model for studying the regulation of eukaryotic metabolism. During the last three decades, the combination of powerful yeast genetics and genome-wide approaches has led to a more integrated view of metabolic regulation. Multiple layers of regulation, from suprapathway control to individual gene responses, have been discovered. Constitutive and dedicated systems that are critical in sensing of the intra- and extracellular environment have been identified, and there is a growing awareness of their involvement in the highly regulated intracellular compartmentalization of proteins and metabolites. This review focuses on recent developments in the field of amino acid, nucleotide, and phosphate metabolism and provides illustrative examples of how yeast cells combine a variety of mechanisms to achieve coordinated regulation of multiple metabolic pathways. Importantly, common schemes have emerged, which reveal mechanisms conserved among various pathways, such as those involved in metabolite sensing and transcriptional regulation by noncoding RNAs or by metabolic intermediates. Thanks to the remarkable sophistication offered by the yeast experimental system, a picture of the intimate connections between the metabolomic and the transcriptome is becoming clear.
Figures
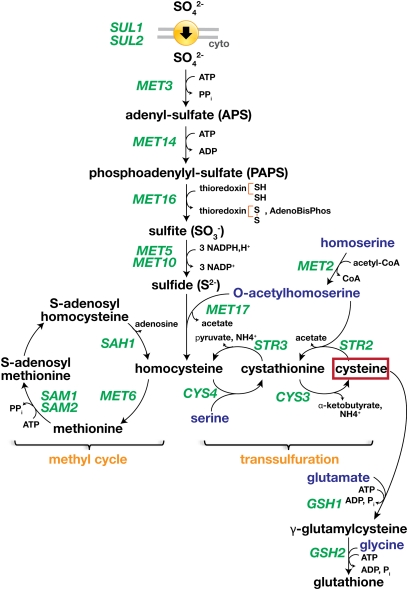
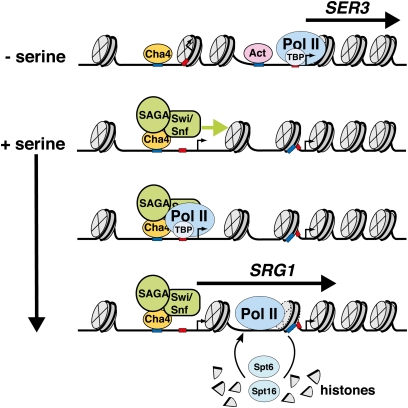
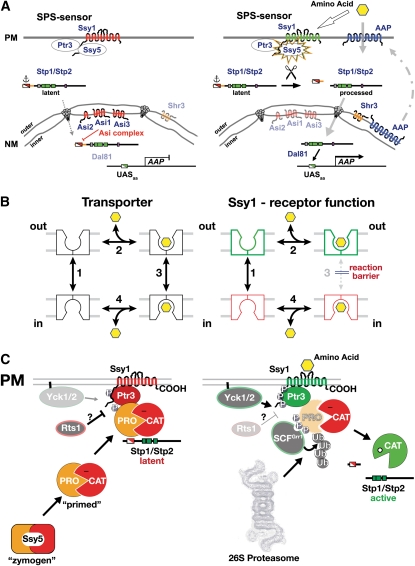
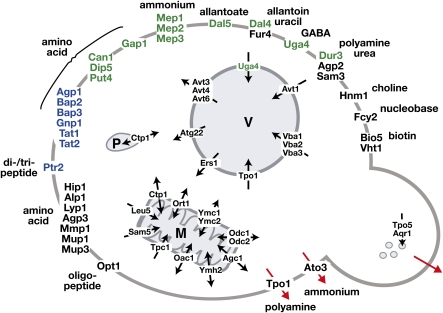
References
-
- Abadjieva A., Hilven P., Pauwels K., Crabeel M., 2000. The yeast ARG7 gene product is autoproteolyzed to two subunit peptides, yielding active ornithine acetyltransferase. J. Biol. Chem. 275: 11361–11367 - PubMed
-
- Abadjieva A., Pauwels K., Hilven P., Crabeel M., 2001. A new yeast metabolon involving at least the two first enzymes of arginine biosynthesis: acetylglutamate synthase activity requires complex formation with acetylglutamate kinase. J. Biol. Chem. 276: 42869–42880 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
