Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration
- PMID: 22421967
- PMCID: PMC3438489
- DOI: 10.1038/cdd.2012.33
Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration
Abstract
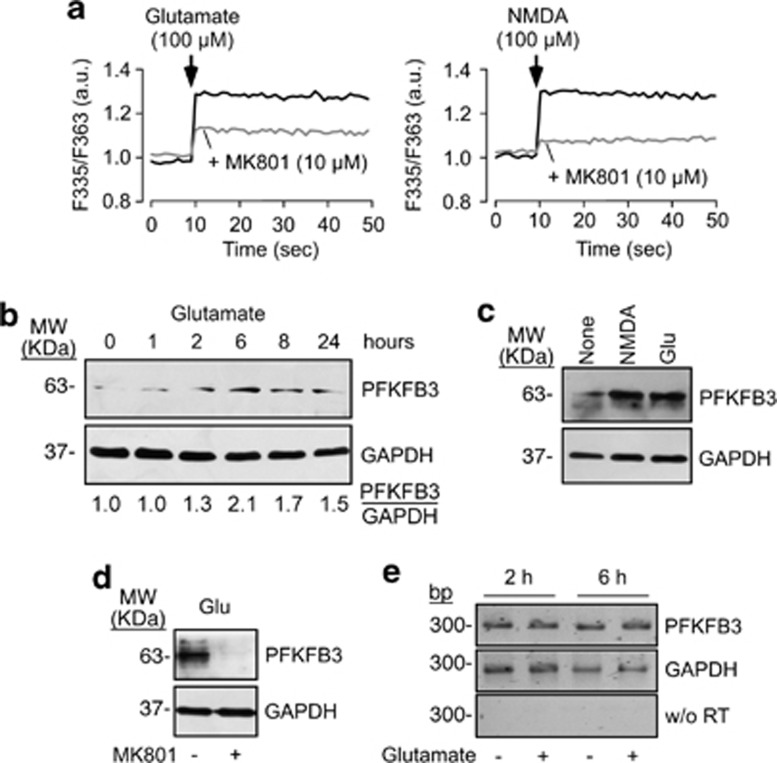
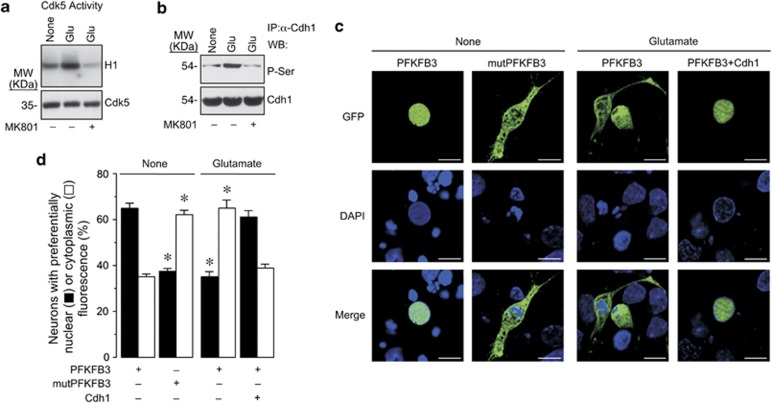
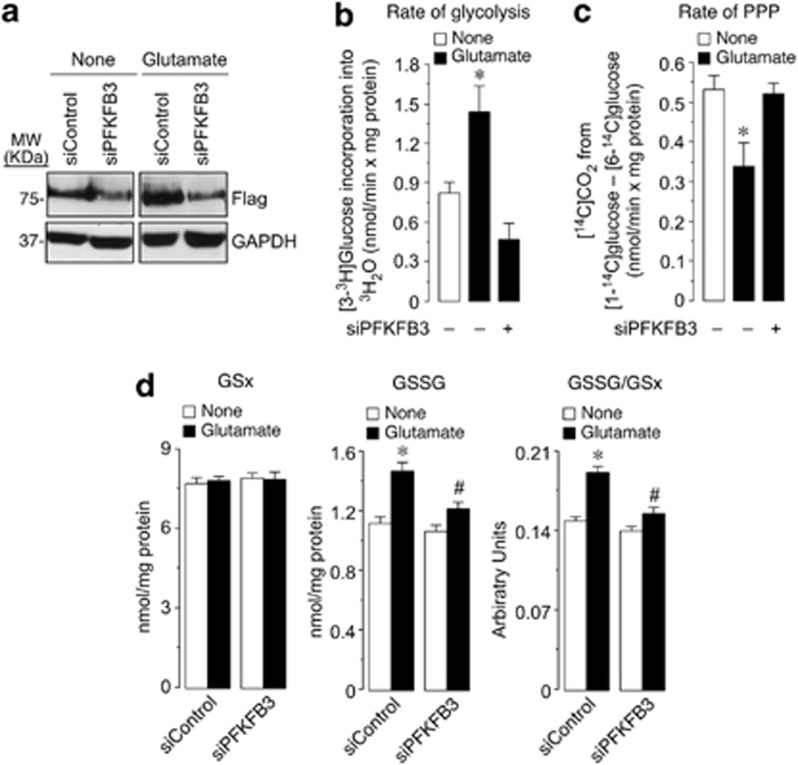
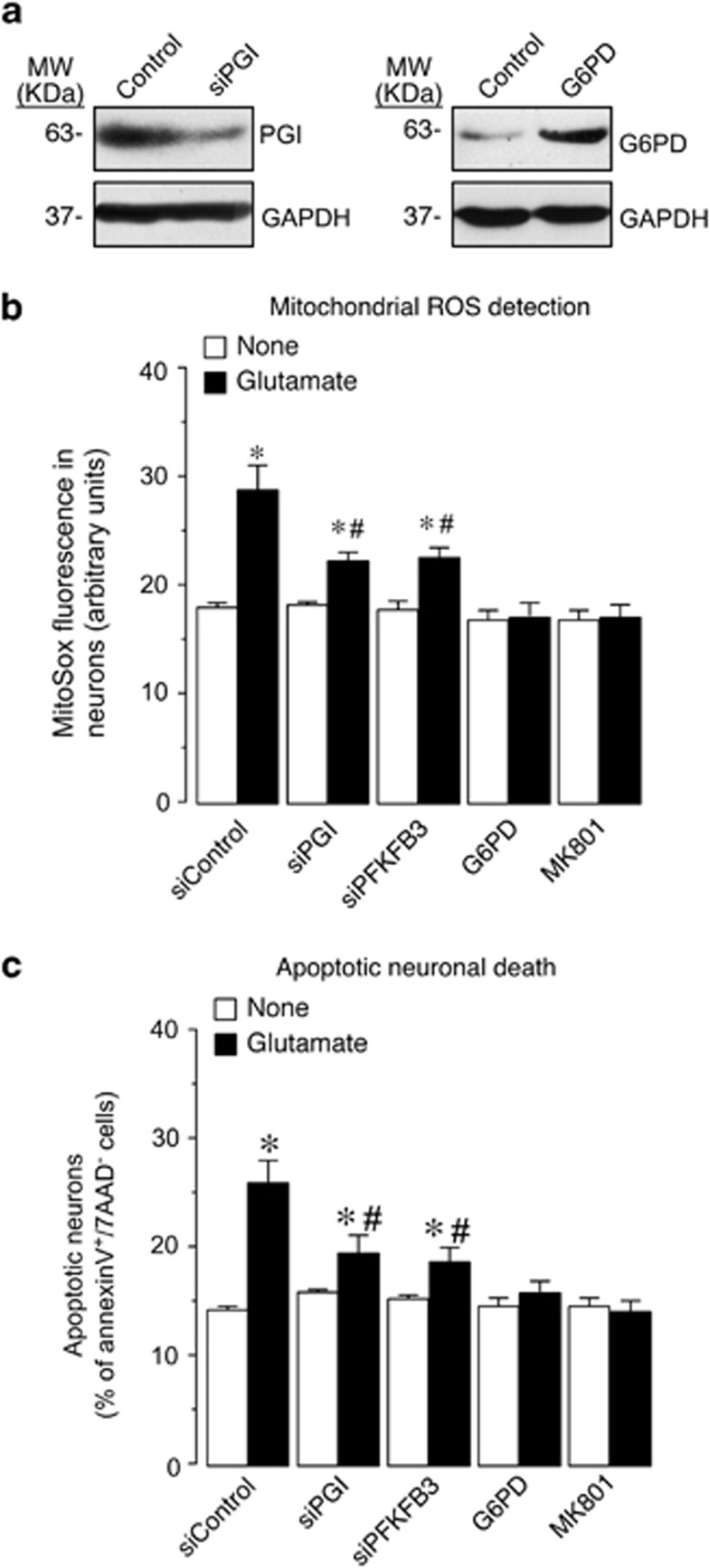
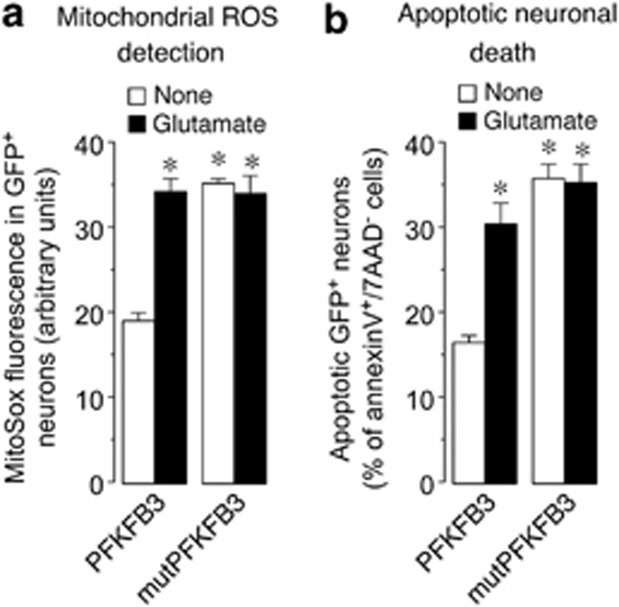
6-Phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) is a master regulator of glycolysis by its ability to synthesize fructose-2,6-bisphosphate, a potent allosteric activator of 6-phosphofructo-1-kinase. Being a substrate of the E3 ubiquitin ligase anaphase-promoting complex-Cdh1 (APC(Cdh1)), PFKFB3 is targeted to proteasomal degradation in neurons. Here, we show that activation of N-methyl-D-aspartate subtype of glutamate receptors (NMDAR) stabilized PFKFB3 protein in cortical neurons. Expressed PFKFB3 was found to be mainly localized in the nucleus, where it is subjected to degradation; however, expression of PFKFB3 lacking the APC(Cdh1)-targeting KEN motif, or following NMDAR stimulation, promoted accumulation of PFKFB3 and its release from the nucleus to the cytosol through an excess Cdh1-inhibitable process. NMDAR-mediated increase in PFKFB3 yielded neurons having a higher glycolysis and lower pentose-phosphate pathway (PPP); this led to oxidative stress and apoptotic neuronal death that was counteracted by overexpressing glucose-6-phosphate dehydrogenase, the rate-limiting enzyme of the PPP. Furthermore, expression of the mutant form of PFKFB3 lacking the KEN motif was sufficient to trigger oxidative stress and apoptotic death of neurons. These results reveal that, by inhibition of APC(Cdh1), glutamate receptors activation stabilizes PFKFB3 thus switching neuronal metabolism leading to oxidative damage and neurodegeneration.
Figures