

Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease
- PMID: 22422451
- PMCID: PMC3304531
- DOI: 10.1093/ije/dyr233
Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease
Abstract
The burgeoning interest in the field of epigenetics has precipitated the need to develop approaches to strengthen causal inference when considering the role of epigenetic mediators of environmental exposures on disease risk. Epigenetic markers, like any other molecular biomarker, are vulnerable to confounding and reverse causation. Here, we present a strategy, based on the well-established framework of Mendelian randomization, to interrogate the causal relationships between exposure, DNA methylation and outcome. The two-step approach first uses a genetic proxy for the exposure of interest to assess the causal relationship between exposure and methylation. A second step then utilizes a genetic proxy for DNA methylation to interrogate the causal relationship between DNA methylation and outcome. The rationale, origins, methodology, advantages and limitations of this novel strategy are presented.
Figures
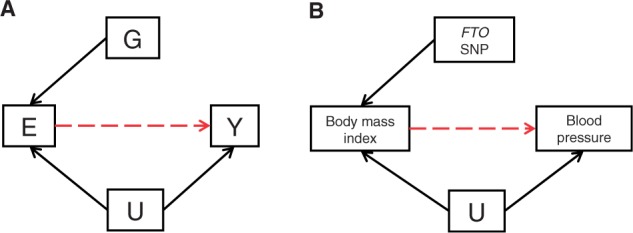

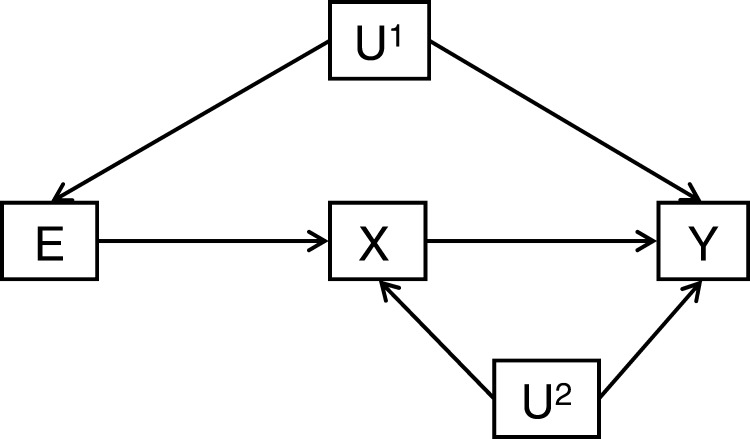
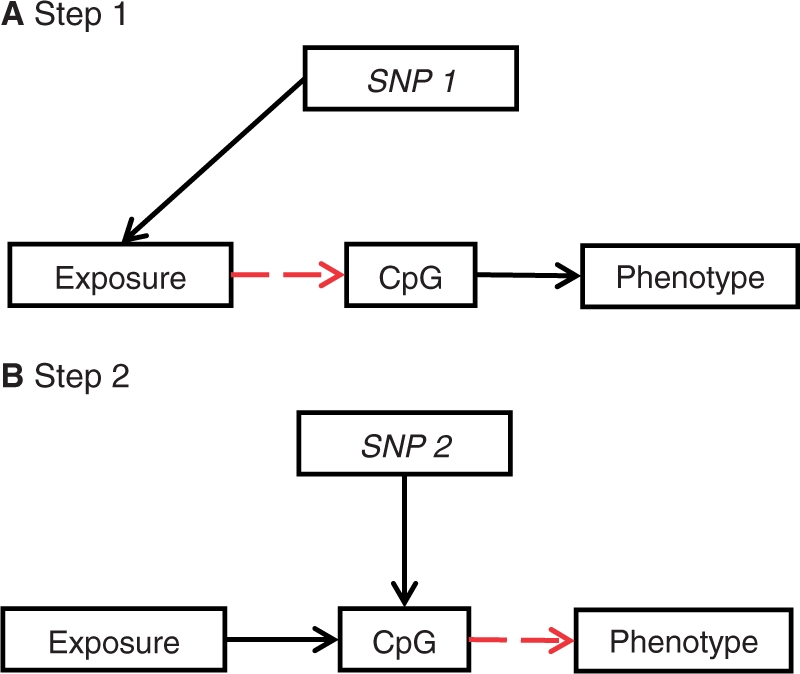
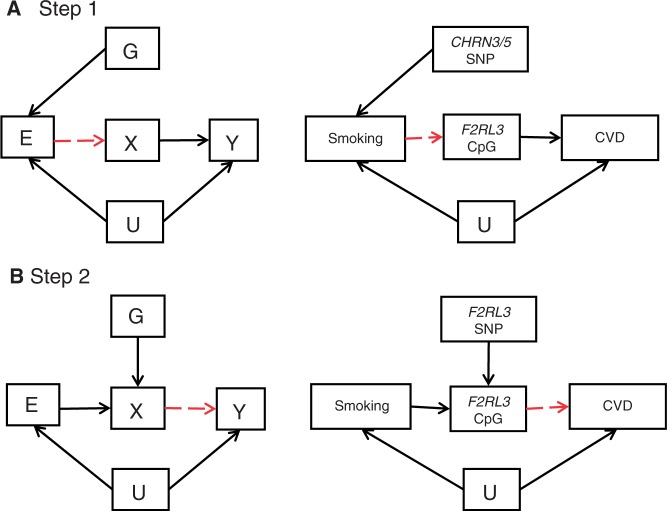
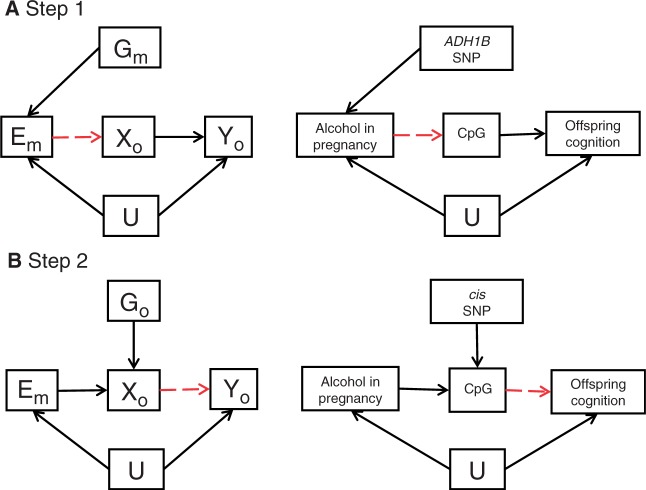
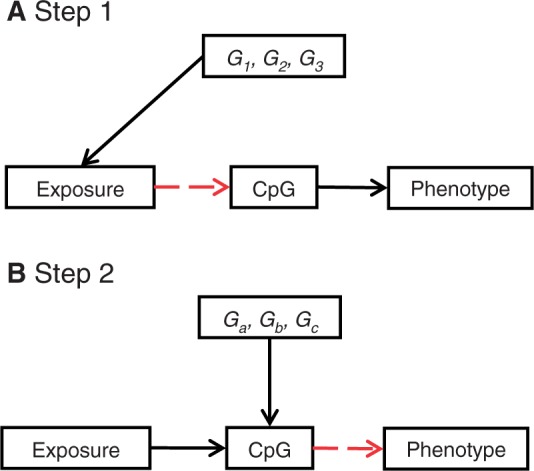
Comment in
-
The education corner: updates on new and established core concepts and methods in epidemiology.Int J Epidemiol. 2012 Apr;41(2):333-4. doi: 10.1093/ije/dys050. Int J Epidemiol. 2012. PMID: 22493321 No abstract available.
References
-
- Hamm CA, Costa FF. The impact of epigenomics on future drug design and new therapies. Drug Discov Today. 2011;16:626–35. - PubMed
-
- Feero WG, Guttmacher AE, Collins FS. Genomic medicine–an updated primer. N Engl J Med. 2010;362:2001–11. - PubMed
-
- Vineis P, Pearce NE. Genome-wide association studies may be misinterpreted: genes versus heritability. Carcinogenesis. 2011;32:1295–98. - PubMed
-
- Ebrahim S, Davey Smith G. Mendelian randomization: can genetic epidemiology help redress the failures of observational epidemiology? Hum Genet. 2008;123:15–33. - PubMed
