

Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy
- PMID: 22422504
- PMCID: PMC3477505
- DOI: 10.1002/hep.25717
Prevention of hepatocellular adenoma and correction of metabolic abnormalities in murine glycogen storage disease type Ia by gene therapy
Abstract
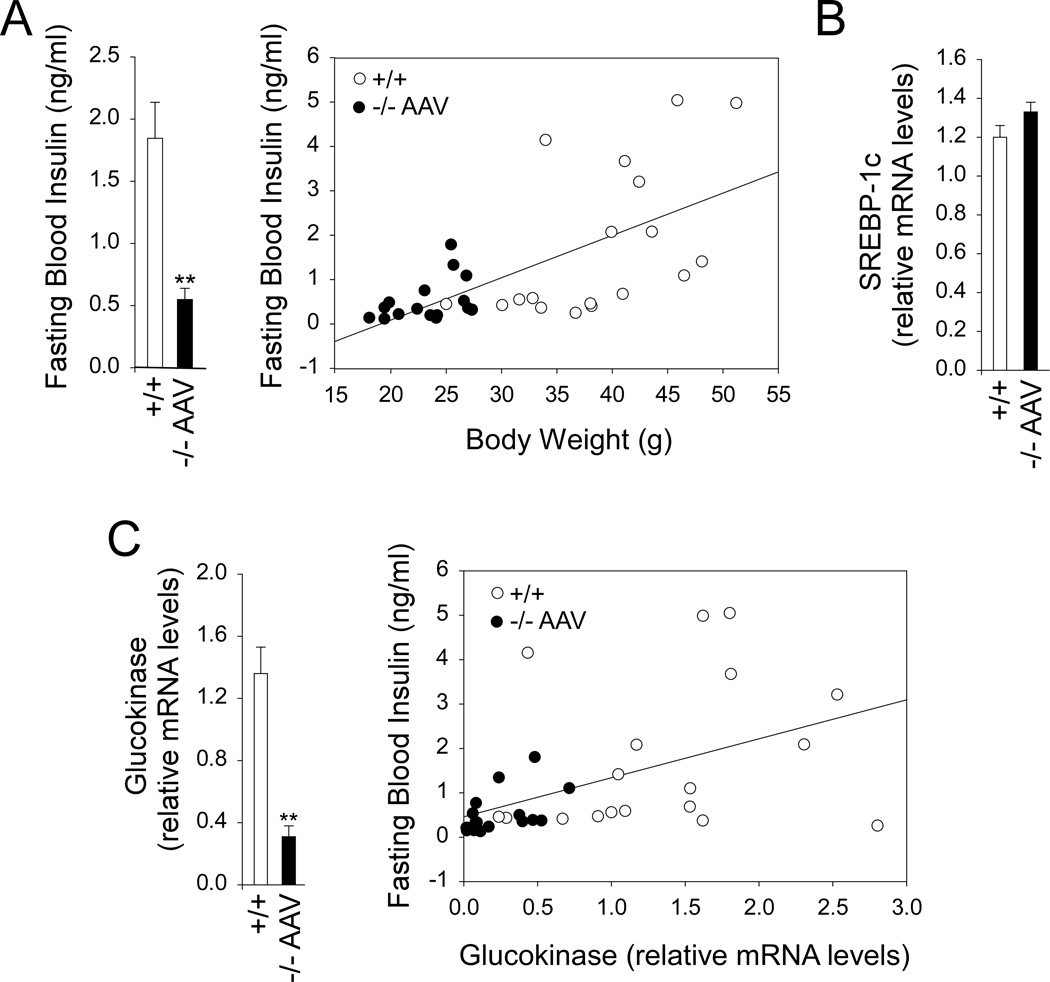
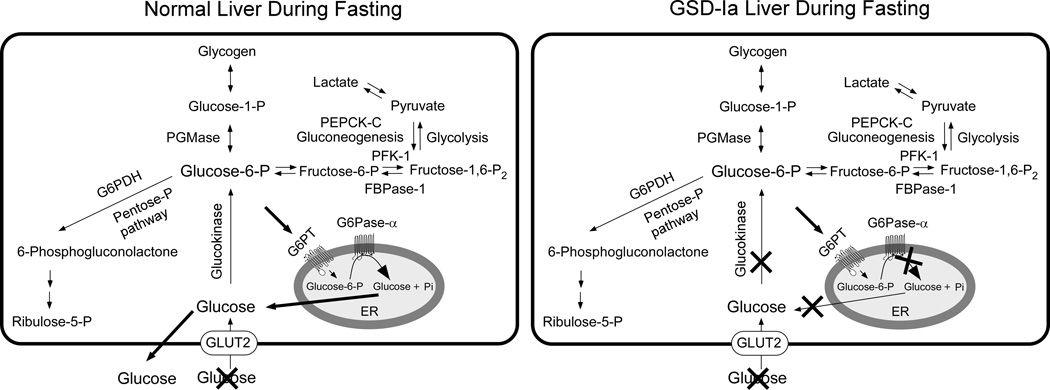
Glycogen storage disease type Ia (GSD-Ia), which is characterized by impaired glucose homeostasis and chronic risk of hepatocellular adenoma (HCA), is caused by deficiencies in the endoplasmic reticulum (ER)-associated glucose-6-phosphatase-α (G6Pase-α or G6PC) that hydrolyzes glucose-6-phosphate (G6P) to glucose. G6Pase-α activity depends on the G6P transporter (G6PT) that translocates G6P from the cytoplasm into the ER lumen. The functional coupling of G6Pase-α and G6PT maintains interprandial glucose homeostasis. We have shown previously that gene therapy mediated by AAV-GPE, an adeno-associated virus (AAV) vector expressing G6Pase-α directed by the human G6PC promoter/enhancer (GPE), completely normalizes hepatic G6Pase-α deficiency in GSD-Ia (G6pc(-/-) ) mice for at least 24 weeks. However, a recent study showed that within 78 weeks of gene deletion, all mice lacking G6Pase-α in the liver develop HCA. We now show that gene therapy mediated by AAV-GPE maintains efficacy for at least 70-90 weeks for mice expressing more than 3% of wild-type hepatic G6Pase-α activity. The treated mice displayed normal hepatic fat storage, had normal blood metabolite and glucose tolerance profiles, had reduced fasting blood insulin levels, maintained normoglycemia over a 24-hour fast, and had no evidence of hepatic abnormalities. After a 24-hour fast, hepatic G6PT messenger RNA levels in G6pc(-/-) mice receiving gene therapy were markedly increased. Because G6PT transport is the rate-limiting step in microsomal G6P metabolism, this may explain why the treated G6pc(-/-) mice could sustain prolonged fasts. The low fasting blood insulin levels and lack of hepatic steatosis may explain the absence of HCA.
Conclusion: These results confirm that AAV-GPE-mediated gene transfer corrects hepatic G6Pase-α deficiency in murine GSD-Ia and prevents chronic HCA formation.
Copyright © 2012 American Association for the Study of Liver Diseases.
Figures





Comment in
-
Adeno-associated virus gene therapy prevents hepatocellular adenoma in murine model of glycogen storage disease type Ia.Hepatology. 2012 Nov;56(5):1593-5. doi: 10.1002/hep.25894. Hepatology. 2012. PMID: 22706804 No abstract available.
References
-
- Greene HL, Slonim AE, O'Neill JA, Jr, Burr IM. Continuous nocturnal intragastric feeding for management of type 1 glycogen-storage disease. N Engl J Med. 1976;294:423–425. - PubMed
-
- Chen YT, Cornblath M, Sidbury JB. Cornstarch therapy in type I glycogen storage disease. N Engl J Med. 1984;310:171–175. - PubMed
-
- Labrune P, Trioche P, Duvaltier I, Chevalier P, Odievre M. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997;24:276–279. - PubMed
-
- Rake JP, Visser G, Labrune P, Leonard JV, Ullrich K, Smit GP. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I) Eur J Pediatr. 2002;161(Suppl 1):S20–S34. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical