

Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations
- PMID: 22422540
- PMCID: PMC3338284
- DOI: 10.2215/CJN.12841211
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis: phenotype-genotype correlation and outcome in 32 patients with CLDN16 or CLDN19 mutations
Abstract
Background and objectives: Familial hypomagnesemia with hypercalciuria and nephrocalcinosis is a rare autosomal recessive renal tubular disease. It is caused by mutations in CLDN16 and CLDN19, encoding claudin-16 and -19, respectively. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis is usually complicated by progressive CKD. The objectives of this study were to describe the clinical and genetic features of familial hypomagnesemia with hypercalciuria and nephrocalcinosis and analyze phenotype-genotype associations in patients with CLDN16 or CLDN19 mutations.
Design, setting, participants, & measurements: Data from 32 genetically confirmed patients (9 patients with CLDN16 and 23 patients with CLDN19 mutations) from 26 unrelated families were retrospectively reviewed.
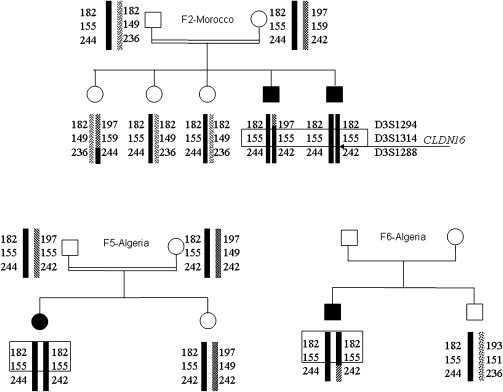
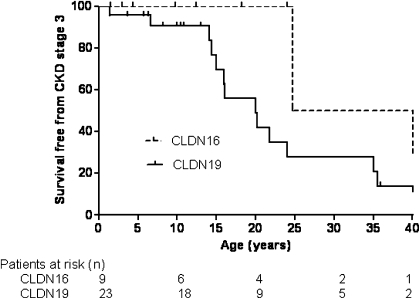
Results: Diagnosis was based on clinical criteria at a median age of 9.5 years and confirmed by genetic testing at a median age of 15.5 years. In total, 13 CLDN16 or CLDN19 mutations were identified, including 8 novel mutations. A founder effect was detected for the recurrent CLDN16 p.Ala139Val mutation in North African families and the CLDN19 p.Gly20Asp mutation in Spanish and French families. CKD was more frequently observed in patients with CLDN19 mutations: survival without CKD or ESRD was 56% at 20 years of age in CLDN19 versus 100% in CLDN16 mutations (log rank P<0.01). Ocular abnormalities were observed in 91% of patients with CLDN19 mutations and none of the patients with CLDN16 mutations (P<0.01). Treatments seem to have no effect on hypercalciuria and CKD progression.
Conclusions: Patients with CLDN19 mutations may display more severe renal impairment than patients with CLDN16 mutations. Ocular abnormalities were observed only in patients with CLDN19 mutations.
Figures

References
-
- Michelis MF, Drash AL, Linarelli LG, De Rubertis FR, Davis BB: Decreased bicarbonate threshold and renal magnesium wasting in a sibship with distal renal tubular acidosis. (Evaluation of the pathophysiological role of parathyroid hormone). Metabolism 21: 905–920, 1972 - PubMed
-
- Konrad M, Hou J, Weber S, Dötsch J, Kari JA, Seeman T, Kuwertz-Bröking E, Peco-Antic A, Tasic V, Dittrich K, Alshaya HO, von Vigier RO, Gallati S, Goodenough DA, Schaller A: CLDN16 genotype predicts renal decline in familial hypomagnesemia with hypercalciuria and nephrocalcinosis. J Am Soc Nephrol 19: 171–181, 2008 - PMC - PubMed
-
- Benigno V, Canonica CS, Bettinelli A, von Vigier RO, Truttmann AC, Bianchetti MG: Hypomagnesaemia-hypercalciuria-nephrocalcinosis: A report of nine cases and a review. Nephrol Dial Transplant 15: 605–610, 2000 - PubMed
-
- Simon DB, Lu Y, Choate KA, Velazquez H, Al-Sabban E, Praga M, Casari G, Bettinelli A, Colussi G, Rodriguez-Soriano J, McCredie D, Milford D, Sanjad S, Lifton RP: Paracellin-1, a renal tight junction protein required for paracellular Mg2+ resorption. Science 285: 103–106, 1999 - PubMed
-
- Konrad M, Schaller A, Seelow D, Pandey AV, Waldegger S, Lesslauer A, Vitzthum H, Suzuki Y, Luk JM, Becker C, Schlingmann KP, Schmid M, Rodriguez-Soriano J, Ariceta G, Cano F, Enriquez R, Juppner H, Bakkaloglu SA, Hediger MA, Gallati S, Neuhauss SC, Nurnberg P, Weber S: Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet 79: 949–957, 2006 - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
