

Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human
- PMID: 22422768
- PMCID: PMC3363337
- DOI: 10.1093/hmg/dds104
Absence of triadin, a protein of the calcium release complex, is responsible for cardiac arrhythmia with sudden death in human
Abstract
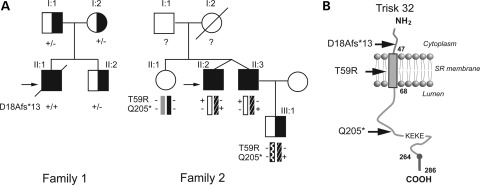
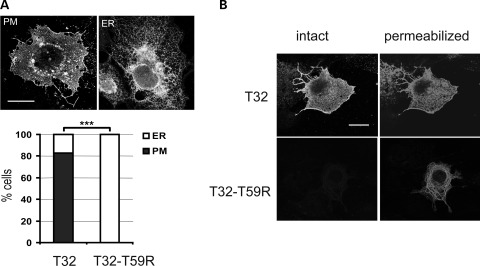
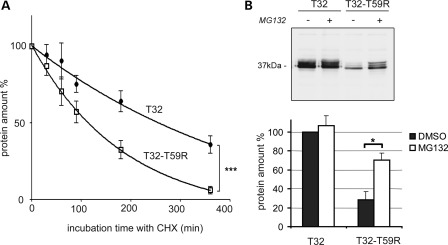
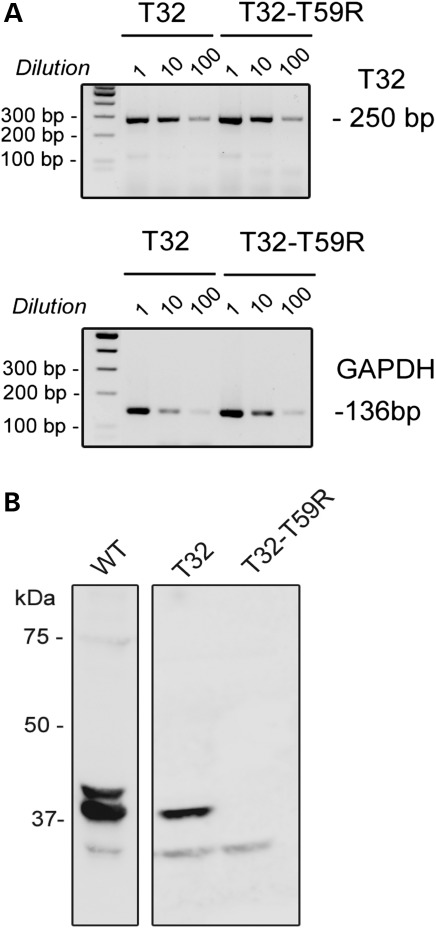
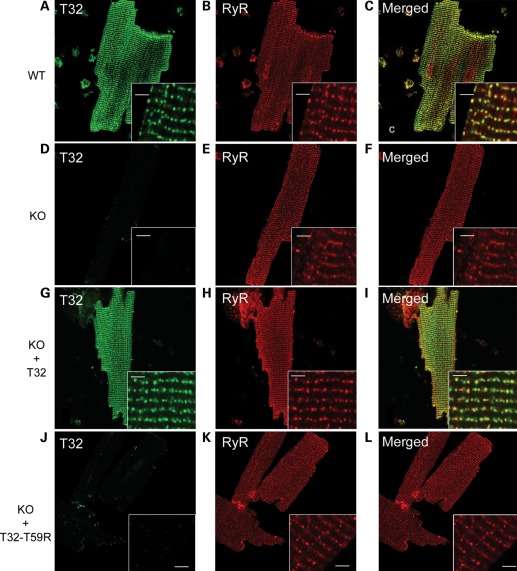
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is an inherited arrhythmogenic disease so far related to mutations in the cardiac ryanodine receptor (RYR2) or the cardiac calsequestrin (CASQ2) genes. Because mutations in RYR2 or in CASQ2 are not retrieved in all CPVT cases, we searched for mutations in the physiological protein partners of RyR2 and CSQ2 in a large cohort of CPVT patients with no detected mutation in these two genes. Based on a candidate gene approach, we focused our investigations on triadin and junctin, two proteins that link RyR2 and CSQ2. Mutations in the triadin (TRDN) and in the junctin (ASPH) genes were searched in a cohort of 97 CPVT patients. We identified three mutations in triadin which cosegregated with the disease on a recessive mode of transmission in two families, but no mutation was found in junctin. Two TRDN mutations, a 4 bp deletion and a nonsense mutation, resulted in premature stop codons; the third mutation, a p.T59R missense mutation, was further studied. Expression of the p.T59R mutant in COS-7 cells resulted in intracellular retention and degradation of the mutant protein. This was confirmed after in vivo expression of the mutant triadin in triadin knock-out mice by viral transduction. In this work, we identified TRDN as a new gene responsible for an autosomal recessive form of CPVT. The mutations identified in the two families lead to the absence of the protein, thereby demonstrating the importance of triadin for the normal function of the cardiac calcium release complex in humans.
Figures
References
-
- Hayashi M., Denjoy I., Extramiana F., Maltret A., Buisson N.R., Lupoglazoff J.M., Klug D., Hayashi M., Takatsuki S., Villain E., et al. Incidence and risk factors of arrhythmic events in catecholaminergic polymorphic ventricular tachycardia. Circulation. 2009;119:2426–2434. doi:10.1161/CIRCULATIONAHA.108.829267. - DOI - PubMed
-
- Priori S.G., Chen S.R. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011;108:871–883. doi:10.1161/CIRCRESAHA.110.226845. - DOI - PMC - PubMed
-
- Guo W., Campbell K.P. Association of triadin with the ryanodine receptor and calsequestrin in the lumen of the sarcoplasmic reticulum. J. Biol. Chem. 1995;270:9027–9030. doi:10.1074/jbc.270.16.9027. - DOI - PubMed
-
- Zhang L., Kelley J., Schmeisser G., Kobayashi Y.M., Jones L.R. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor. Proteins of the cardiac junctional sarcoplasmic reticulum membrane. J. Biol. Chem. 1997;272:23389–23397. doi:10.1074/jbc.272.37.23389. - DOI - PubMed
-
- Thevenon D., Smida-Rezgui S., Chevessier F., Groh S., Henry-Berger J., Romero N.B., Villaz M., De Waard M., Marty I. Human skeletal muscle triadin: gene organization and cloning of the major isoform, Trisk 51. Biochem. Biophys. Res. Commun. 2003;303:669–675. doi:10.1016/S0006-291X(03)00406-6. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
