

Genetics of human cardiovascular disease
- PMID: 22424232
- PMCID: PMC3319439
- DOI: 10.1016/j.cell.2012.03.001
Genetics of human cardiovascular disease
Abstract
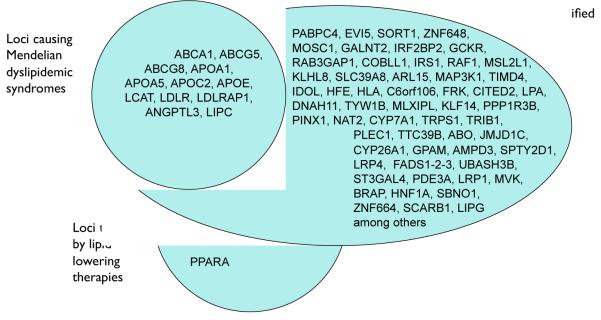
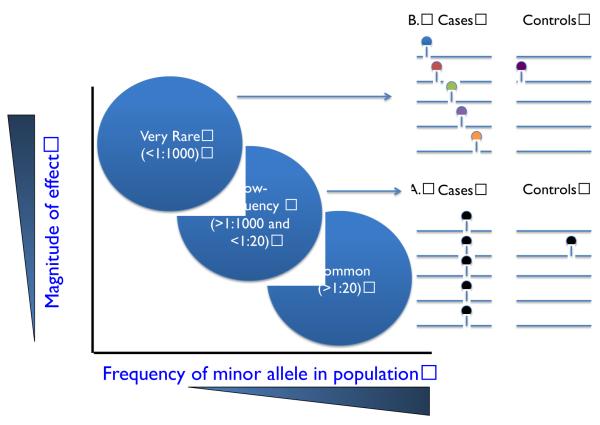
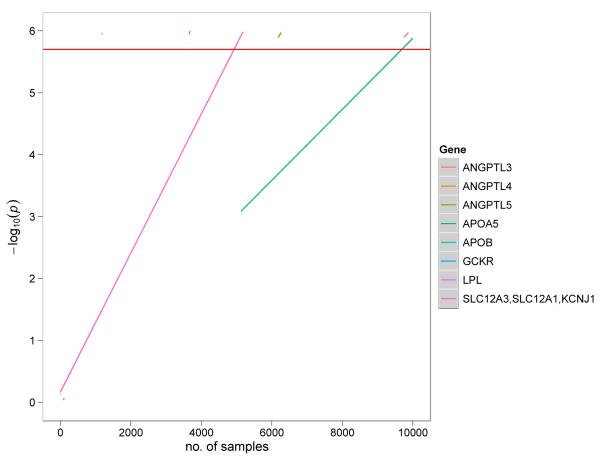
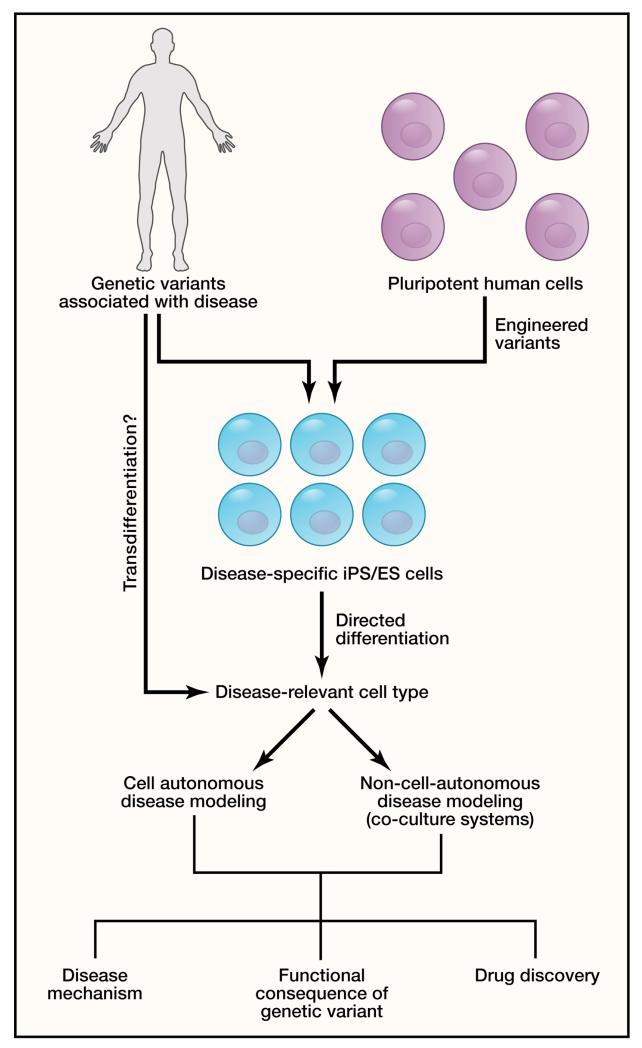
Cardiovascular disease encompasses a range of conditions extending from myocardial infarction to congenital heart disease, most of which are heritable. Enormous effort has been invested in understanding the genes and specific DNA sequence variants that are responsible for this heritability. Here, we review the lessons learned for monogenic and common, complex forms of cardiovascular disease. We also discuss key challenges that remain for gene discovery and for moving from genomic localization to mechanistic insights, with an emphasis on the impact of next-generation sequencing and the use of pluripotent human cells to understand the mechanism by which genetic variation contributes to disease.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
References
-
- Abifadel M, Varret M, Rabes JP, Allard D, Ouguerram K, Devillers M, Cruaud C, Benjannet S, Wickham L, Erlich D, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–156. - PubMed
-
- Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, Shendure J. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–755. - PubMed
-
- Basson CT, Bachinsky DR, Lin RC, Levi T, Elkins JA, Soults J, Grayzel D, Kroumpouzou E, Traill TA, Leblanc-Straceski J, et al. Mutations in human TBX5 [corrected] cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997;15:30–35. - PubMed
-
- Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290:1771–1775. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
