

Mutations in ROGDI Cause Kohlschütter-Tönz Syndrome
- PMID: 22424600
- PMCID: PMC3322220
- DOI: 10.1016/j.ajhg.2012.02.012
Mutations in ROGDI Cause Kohlschütter-Tönz Syndrome
Abstract
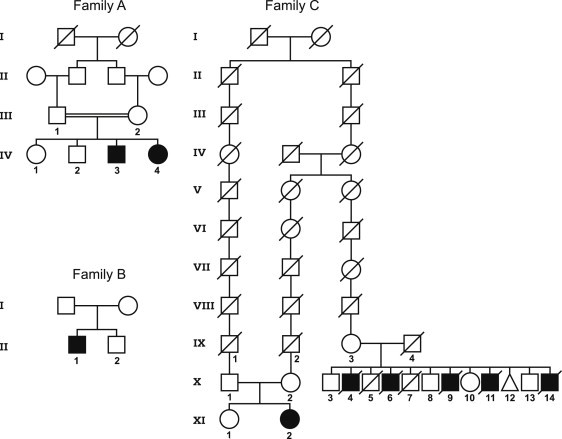
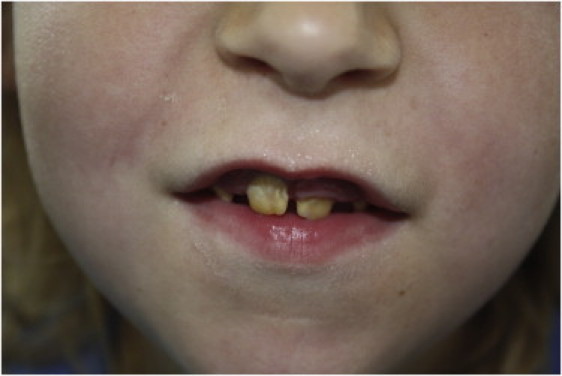
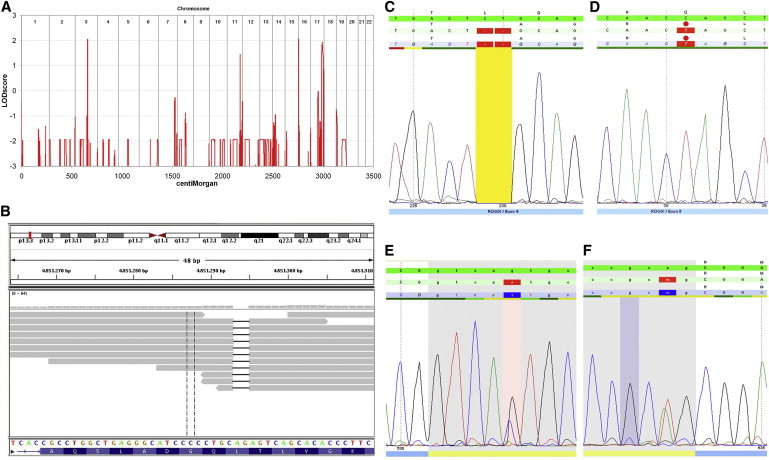
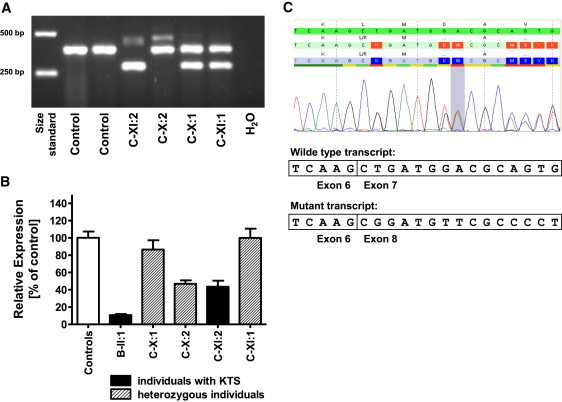
Kohlschütter-Tönz syndrome (KTS) is an autosomal-recessive disease characterized by the combination of epilepsy, psychomotor regression, and amelogenesis imperfecta. The molecular basis has not yet been elucidated. Here, we report that KTS is caused by mutations in ROGDI. Using a combination of autozygosity mapping and exome sequencing, we identified a homozygous frameshift deletion, c.229_230del (p.Leu77Alafs(∗)64), in ROGDI in two affected individuals from a consanguineous family. Molecular studies in two additional KTS-affected individuals from two unrelated Austrian and Swiss families revealed homozygosity for nonsense mutation c.286C>T (p.Gln96(∗)) and compound heterozygosity for the splice-site mutations c.531+5G>C and c.532-2A>T in ROGDI, respectively. The latter mutation was also found to be heterozygous in the mother of the Swiss affected individual in whom KTS was reported for the first time in 1974. ROGDI is highly expressed throughout the brain and other organs, but its function is largely unknown. Possible interactions with DISC1, a protein involved in diverse cytoskeletal functions, have been suggested. Our finding that ROGDI mutations cause KTS indicates that the protein product of this gene plays an important role in neuronal development as well as amelogenesis.
Copyright © 2012 The American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Kohlschütter A., Chappuis D., Meier C., Tönz O., Vassella F., Herschkowitz N. Familial epilepsy and yellow teeth—a disease of the CNS associated with enamel hypoplasia. Helv. Paediatr. Acta. 1974;29:283–294. - PubMed
-
- Petermöller M., Kunze J., Gross-Selbeck G. Kohlschütter syndrome: Syndrome of epilepsy—dementia—amelogenesis imperfecta. Neuropediatrics. 1993;24:337–338. - PubMed
-
- Zlotogora J., Fuks A., Borochowitz Z., Tal Y. Kohlschütter-Tönz syndrome: Epilepsy, dementia, and amelogenesis imperfecta. Am. J. Med. Genet. 1993;46:453–454. - PubMed
-
- Musumeci S.A., Elia M., Ferri R., Romano C., Scuderi C., Del Gracco S. A further family with epilepsy, dementia and yellow teeth: The Kohlschütter syndrome. Brain Dev. 1995;17:133–138. discussion 142–133. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
