

Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation
- PMID: 22426077
- PMCID: PMC3717580
- DOI: 10.1038/ncb2466
Telomeric DNA damage is irreparable and causes persistent DNA-damage-response activation
Erratum in
- Nat Cell Biol. 2012 May;14(5):555
Abstract
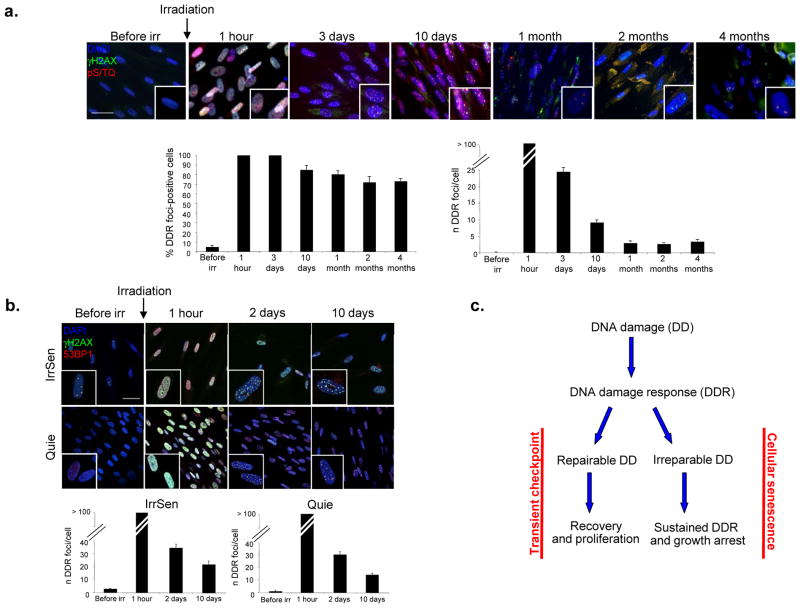
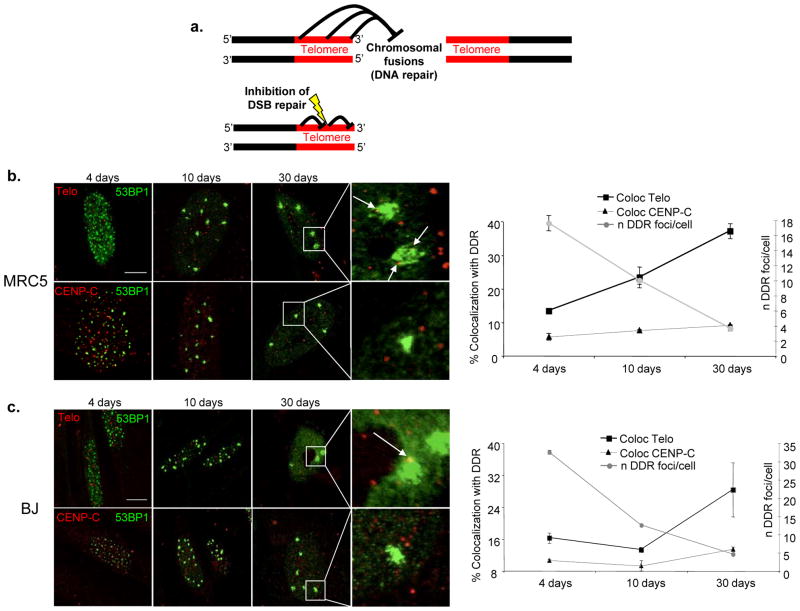
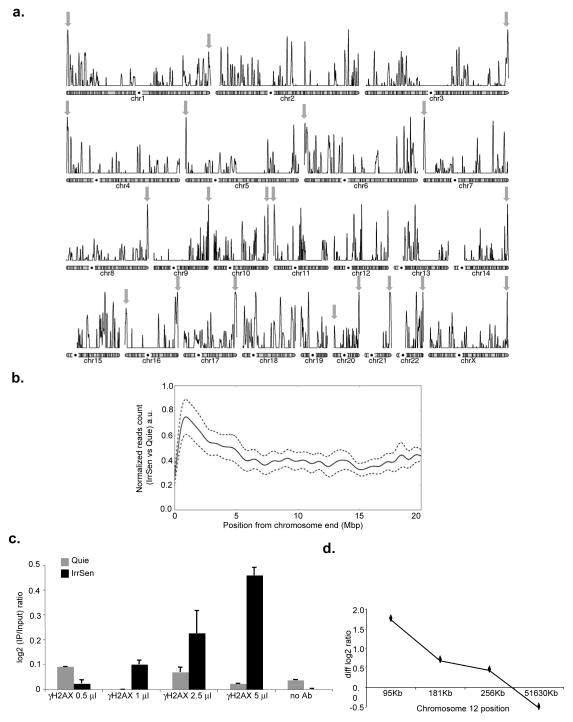
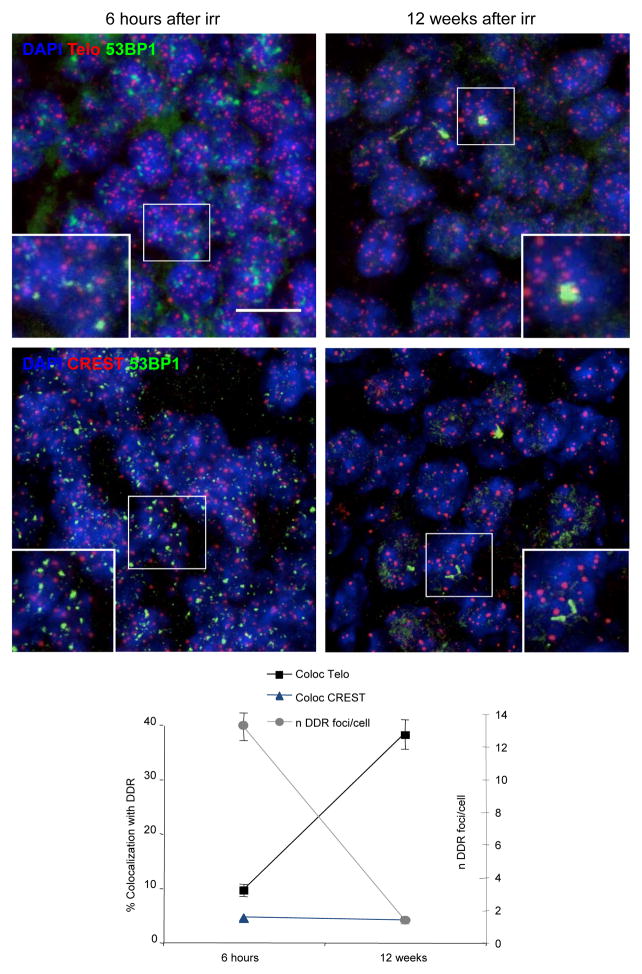
The DNA-damage response (DDR) arrests cell-cycle progression until damage is removed. DNA-damage-induced cellular senescence is associated with persistent DDR. The molecular bases that distinguish transient from persistent DDR are unknown. Here we show that a large fraction of exogenously induced persistent DDR markers is associated with telomeric DNA in cultured cells and mammalian tissues. In yeast, a chromosomal DNA double-strand break next to a telomeric sequence resists repair and impairs DNA ligase 4 recruitment. In mammalian cells, ectopic localization of telomeric factor TRF2 next to a double-strand break induces persistent DNA damage and DDR. Linear, but not circular, telomeric DNA or scrambled DNA induces a prolonged checkpoint in normal cells. In terminally differentiated tissues of old primates, DDR markers accumulate at telomeres that are not critically short. We propose that linear genomes are not uniformly reparable and that telomeric DNA tracts, if damaged, are irreparable and trigger persistent DDR and cellular senescence.
Conflict of interest statement
The authors declare no conflict of interests.
Figures
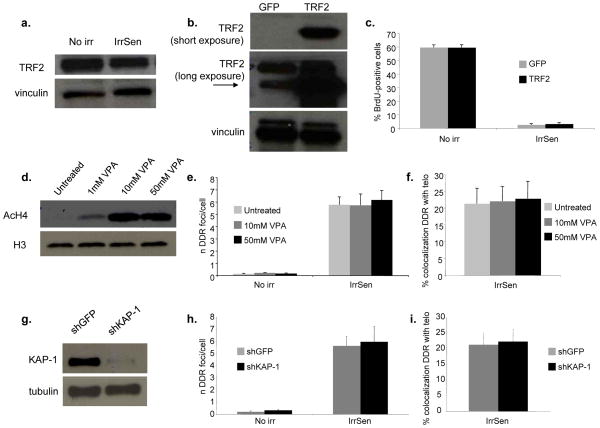
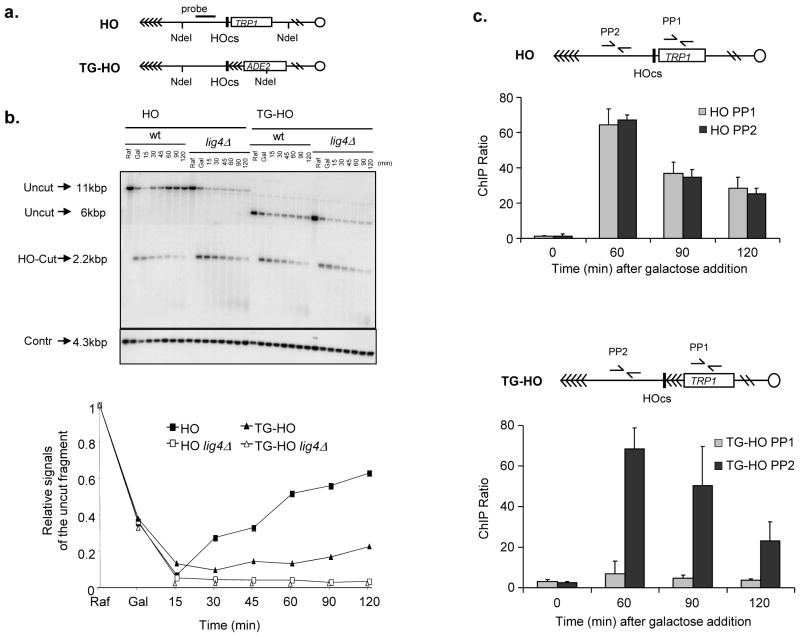
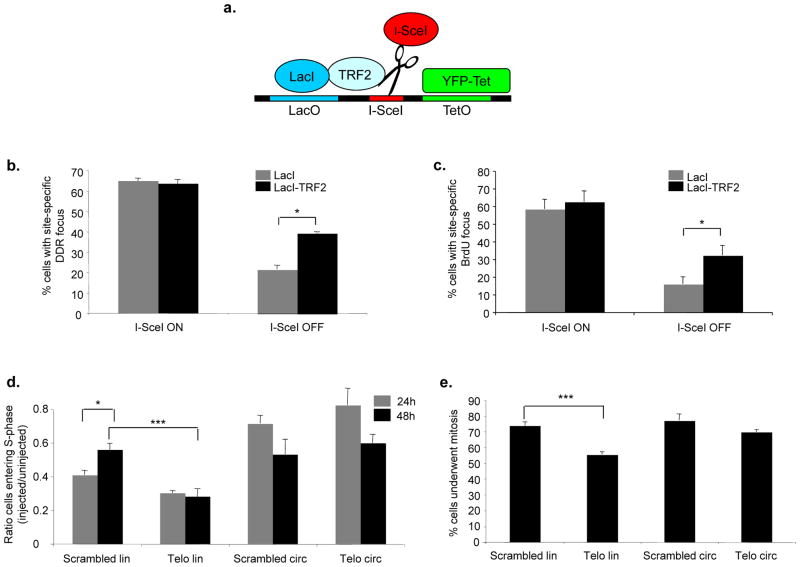
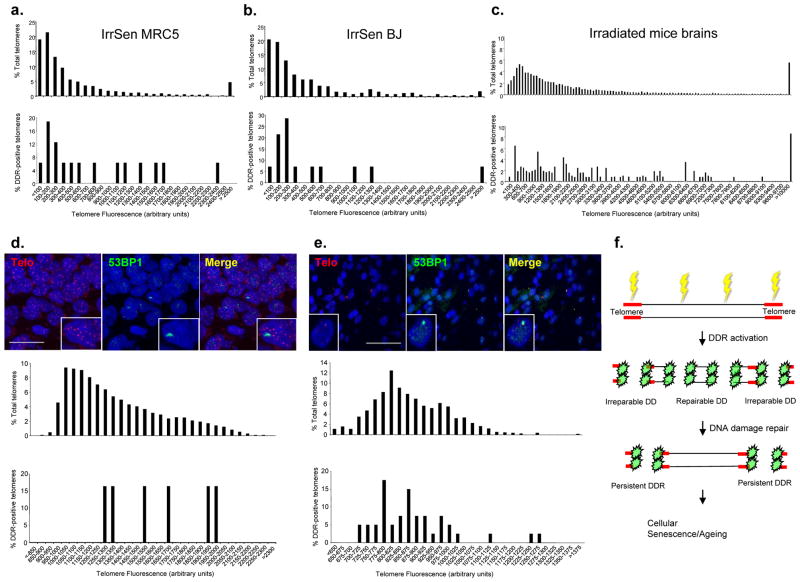
Comment in
-
Telomeres: The perils of peripheral damage.Nat Rev Mol Cell Biol. 2012 Mar 22;13(4):208-9. doi: 10.1038/nrm3318. Nat Rev Mol Cell Biol. 2012. PMID: 22436739 No abstract available.
-
Signalling the end of the line.Nat Cell Biol. 2012 Apr 2;14(4):339-41. doi: 10.1038/ncb2476. Nat Cell Biol. 2012. PMID: 22469830
References
-
- Campisi J, d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. - PubMed
-
- Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. - PubMed
-
- Braig M, et al. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. - PubMed
-
- Collado M, et al. Tumour biology: senescence in premalignant tumours. Nature. 2005;436:642. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
