

Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease
- PMID: 22426213
- PMCID: PMC3314469
- DOI: 10.1172/JCI61134
Mouse and computational models link Mlc2v dephosphorylation to altered myosin kinetics in early cardiac disease
Abstract
Actin-myosin interactions provide the driving force underlying each heartbeat. The current view is that actin-bound regulatory proteins play a dominant role in the activation of calcium-dependent cardiac muscle contraction. In contrast, the relevance and nature of regulation by myosin regulatory proteins (for example, myosin light chain-2 [MLC2]) in cardiac muscle remain poorly understood. By integrating gene-targeted mouse and computational models, we have identified an indispensable role for ventricular Mlc2 (Mlc2v) phosphorylation in regulating cardiac muscle contraction. Cardiac myosin cycling kinetics, which directly control actin-myosin interactions, were directly affected, but surprisingly, Mlc2v phosphorylation also fed back to cooperatively influence calcium-dependent activation of the thin filament. Loss of these mechanisms produced early defects in the rate of cardiac muscle twitch relaxation and ventricular torsion. Strikingly, these defects preceded the left ventricular dysfunction of heart disease and failure in a mouse model with nonphosphorylatable Mlc2v. Thus, there is a direct and early role for Mlc2 phosphorylation in regulating actin-myosin interactions in striated muscle contraction, and dephosphorylation of Mlc2 or loss of these mechanisms can play a critical role in heart failure.
Figures
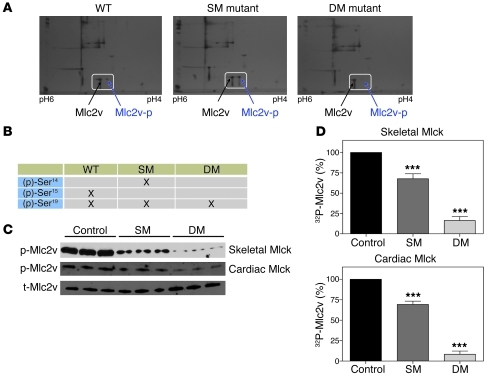
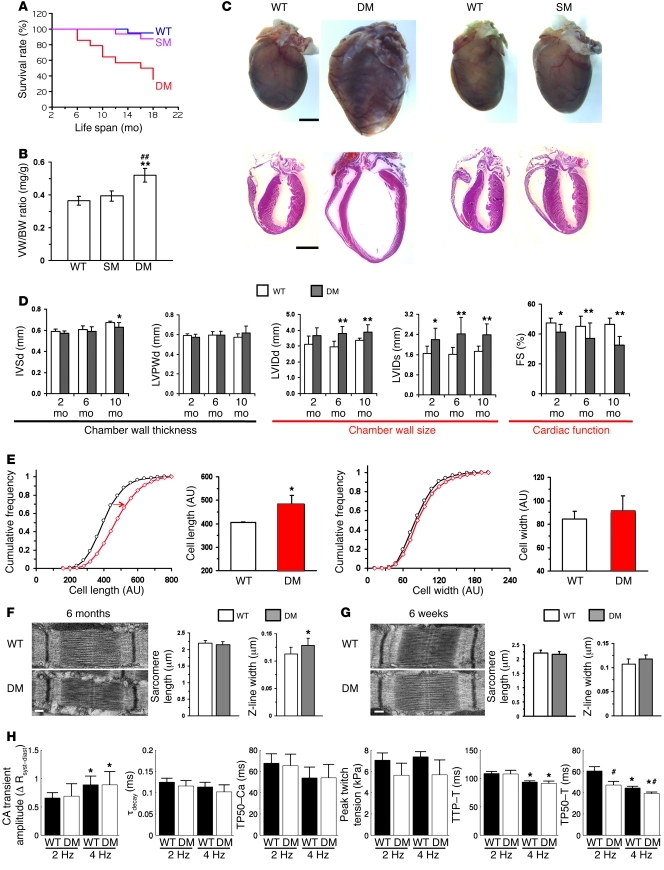
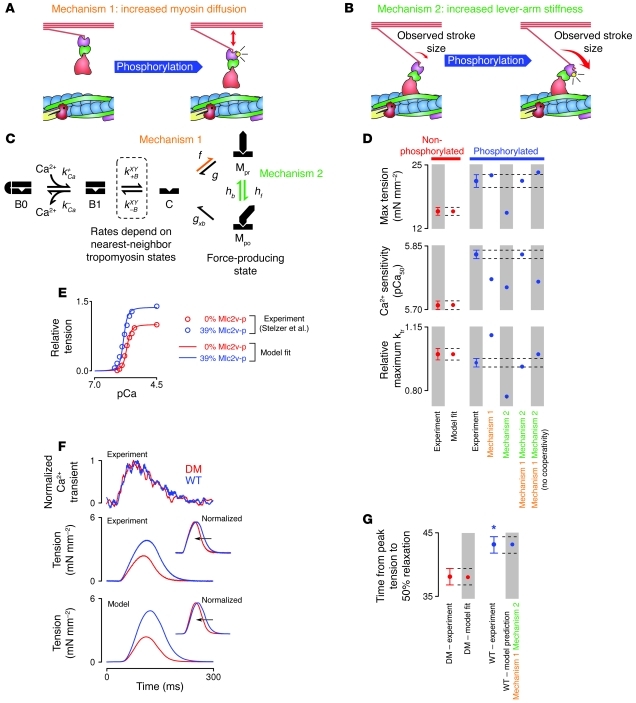
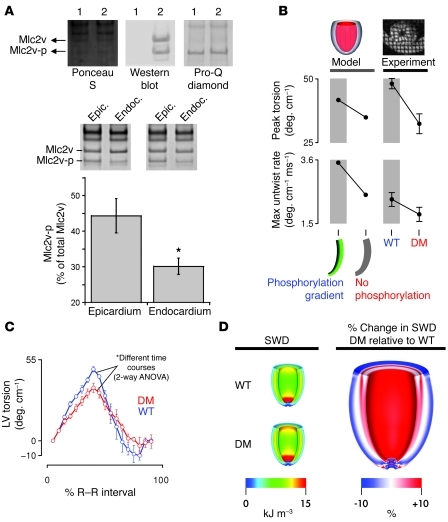
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
