

Chaperone insufficiency links TLR4 protein signaling to endoplasmic reticulum stress
- PMID: 22427667
- PMCID: PMC3346100
- DOI: 10.1074/jbc.M111.315218
Chaperone insufficiency links TLR4 protein signaling to endoplasmic reticulum stress
Retraction in
-
Chaperone insufficiency links TLR4 signaling to endoplasmic reticulum stress.J Biol Chem. 2016 Dec 23;291(52):26936. doi: 10.1074/jbc.A111.315218. J Biol Chem. 2016. PMID: 28011878 Free PMC article. No abstract available.
Abstract
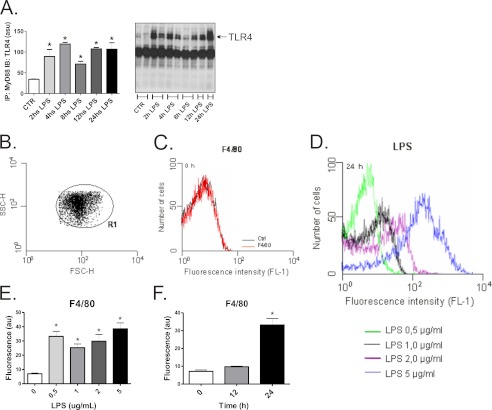
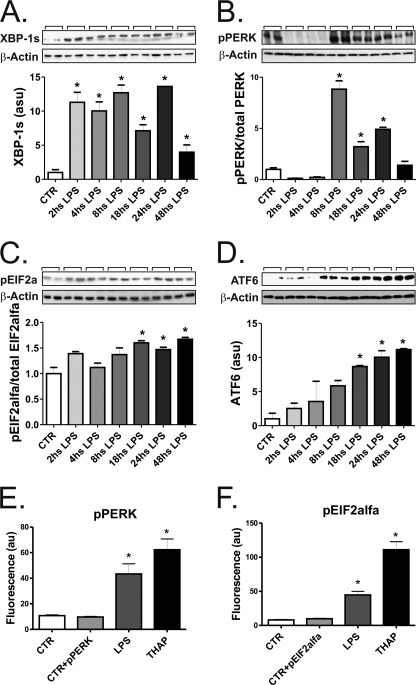
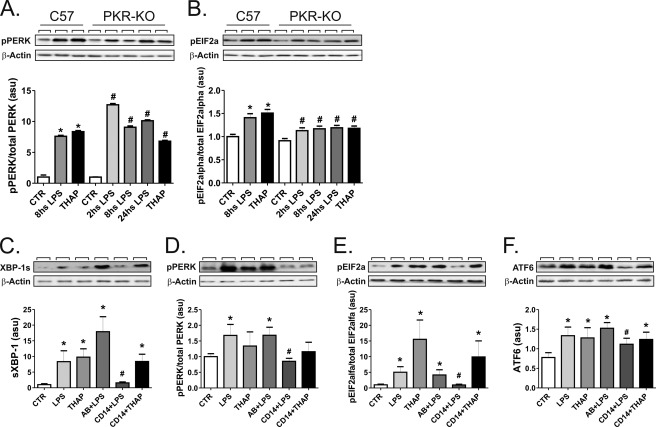
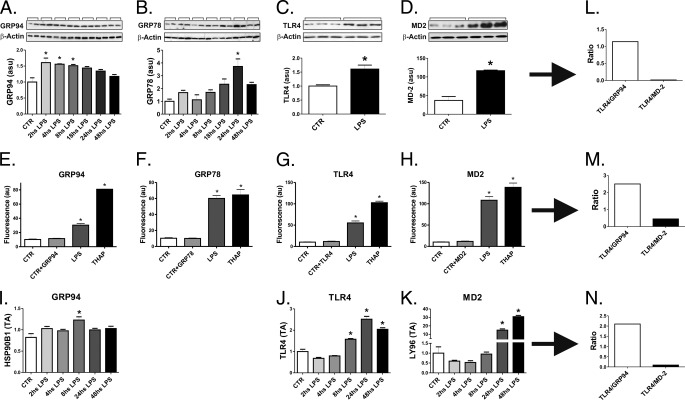
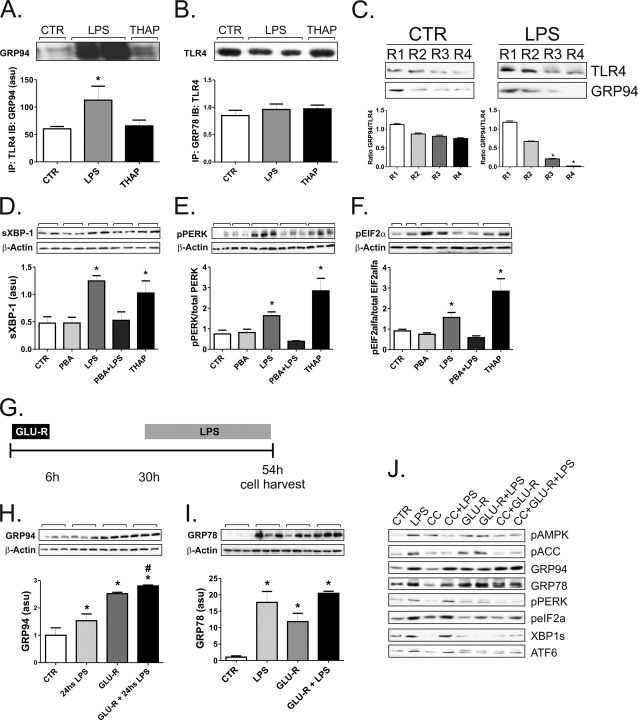
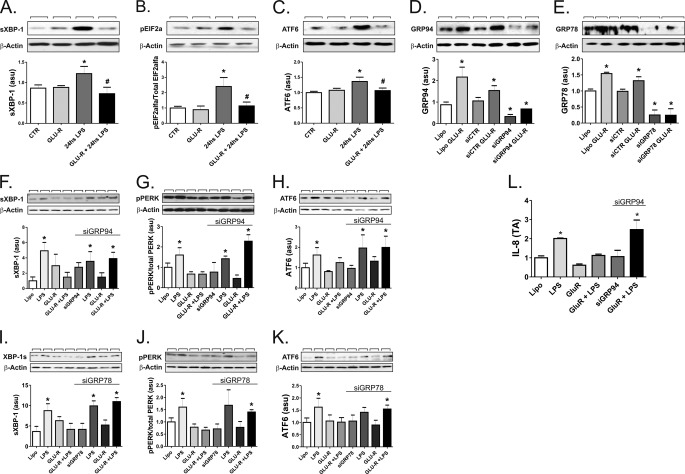
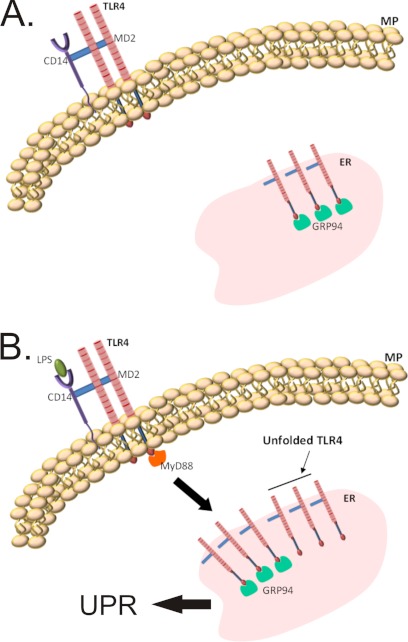
Inflammation plays an important pathogenic role in a number of metabolic diseases such as obesity, type 2 diabetes, and atherosclerosis. The activation of inflammation in these diseases depends at least in part on the combined actions of TLR4 signaling and endoplasmic reticulum stress, which by acting in concert can boost the inflammatory response. Defining the mechanisms involved in this phenomenon may unveil potential targets for the treatment of metabolic/inflammatory diseases. Here we used LPS to induce endoplasmic reticulum stress in the human monocyte cell-line, THP-1. The unfolded protein response, produced after LPS, was dependent on CD14 activity but not on RNA-dependent protein kinase and could be inhibited by an exogenous chemical chaperone. The induction of the endoplasmic reticulum resident chaperones, GRP94 and GRP78, by LPS was of a much lower magnitude than the effect of LPS on TLR4 and MD-2 expression. In face of this apparent insufficiency of chaperone expression, we induced the expression of GRP94 and GRP78 by glucose deprivation. This approach completely reverted endoplasmic reticulum stress. The inhibition of either GRP94 or GRP78 with siRNA was sufficient to rescue the protective effect of glucose deprivation on LPS-induced endoplasmic reticulum stress. Thus, insufficient LPS-induced chaperone expression links TLR4 signaling to endoplasmic reticulum stress.
Figures
References
-
- Gregor M. F., Hotamisligil G. S. (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29, 415–445 - PubMed
-
- Tsukumo D. M., Carvalho-Filho M. A., Carvalheira J. B., Prada P. O., Hirabara S. M., Schenka A. A., Araújo E. P., Vassallo J., Curi R., Velloso L. A., Saad M. J. (2007) Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes 56, 1986–1998 - PubMed
-
- Ozcan U., Cao Q., Yilmaz E., Lee A. H., Iwakoshi N. N., Ozdelen E., Tuncman G., Görgün C., Glimcher L. H., Hotamisligil G. S. (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306, 457–461 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
