

Genetic architecture of highly complex chemical resistance traits across four yeast strains
- PMID: 22438822
- PMCID: PMC3305394
- DOI: 10.1371/journal.pgen.1002570
Genetic architecture of highly complex chemical resistance traits across four yeast strains
Abstract
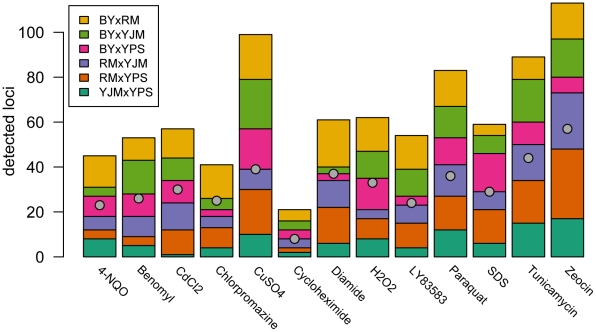
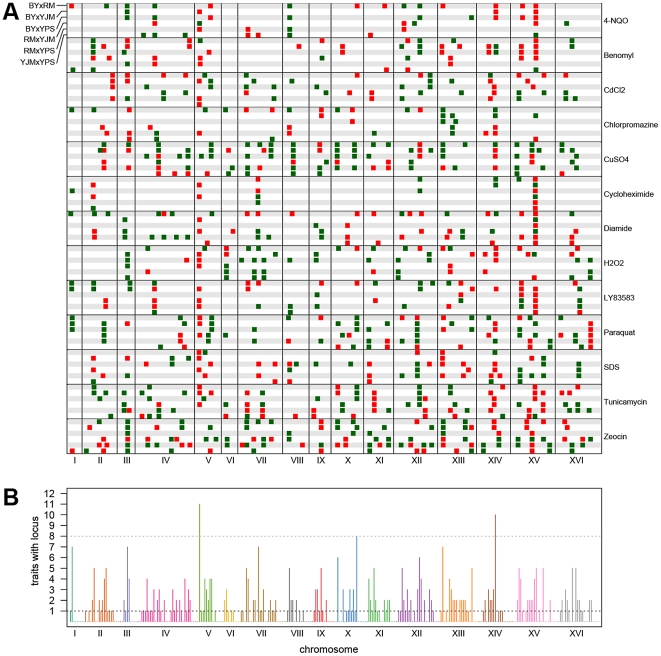
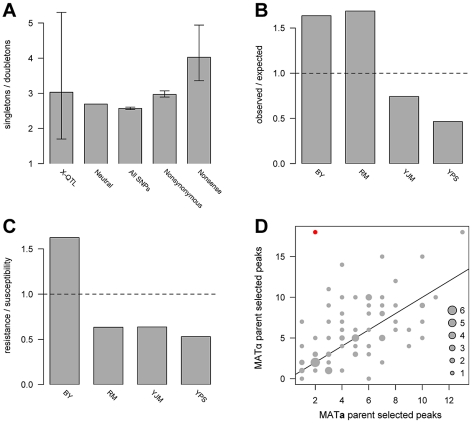
Many questions about the genetic basis of complex traits remain unanswered. This is in part due to the low statistical power of traditional genetic mapping studies. We used a statistically powerful approach, extreme QTL mapping (X-QTL), to identify the genetic basis of resistance to 13 chemicals in all 6 pairwise crosses of four ecologically and genetically diverse yeast strains, and we detected a total of more than 800 loci. We found that the number of loci detected in each experiment was primarily a function of the trait (explaining 46% of the variance) rather than the cross (11%), suggesting that the level of genetic complexity is a consistent property of a trait across different genetic backgrounds. Further, we observed that most loci had trait-specific effects, although a small number of loci with effects in many conditions were identified. We used the patterns of resistance and susceptibility alleles in the four parent strains to make inferences about the allele frequency spectrum of functional variants. We also observed evidence of more complex allelic series at a number of loci, as well as strain-specific signatures of selection. These results improve our understanding of complex traits in yeast and have implications for study design in other organisms.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Falconer DS, Mackay TF. Introduction to quantitative genetics (4th edition) Harlow, England: Pearson Education Limited; 1996.
-
- Mackay TF, Stone EA, Ayroles JF. The genetics of quantitative traits: challenges and prospects. Nat Rev Genet. 2009;10:565–577. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
