

Temporal transcriptional profiling of somatic and germ cells reveals biased lineage priming of sexual fate in the fetal mouse gonad
- PMID: 22438826
- PMCID: PMC3305395
- DOI: 10.1371/journal.pgen.1002575
Temporal transcriptional profiling of somatic and germ cells reveals biased lineage priming of sexual fate in the fetal mouse gonad
Abstract
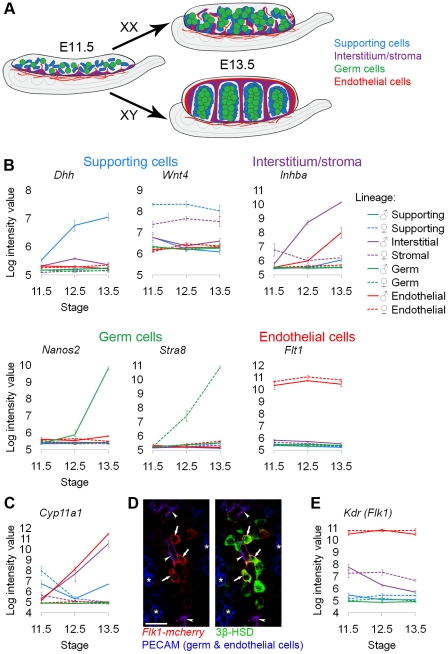
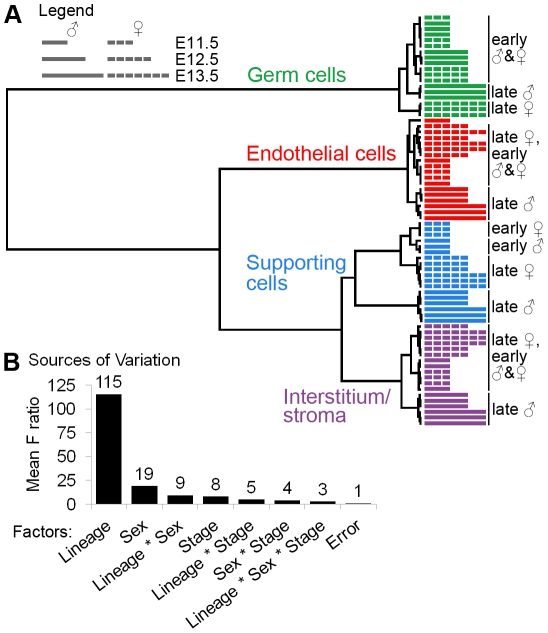
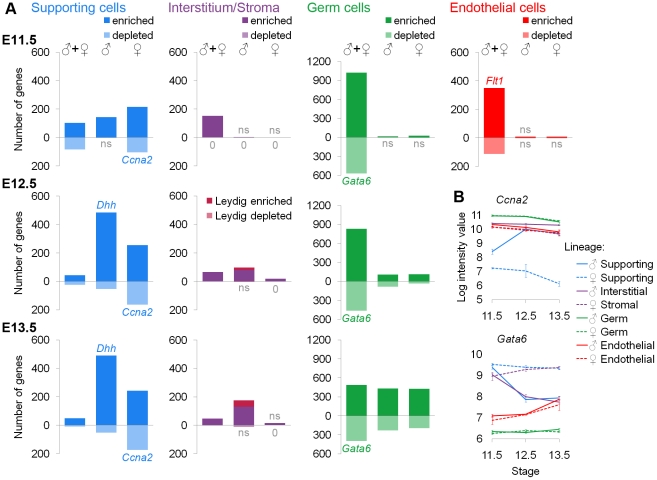
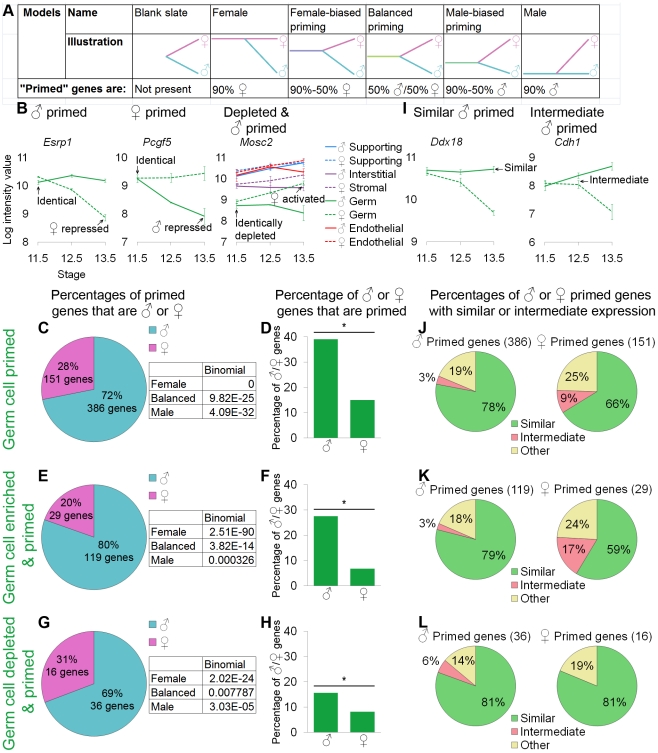
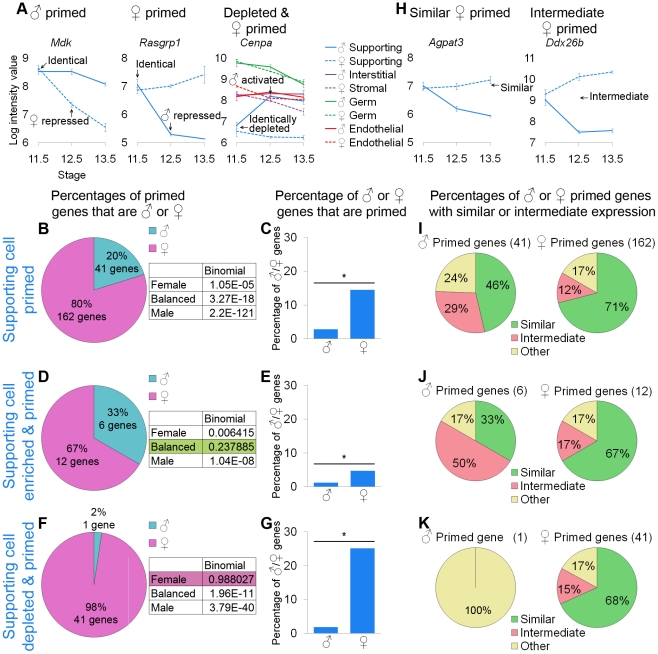
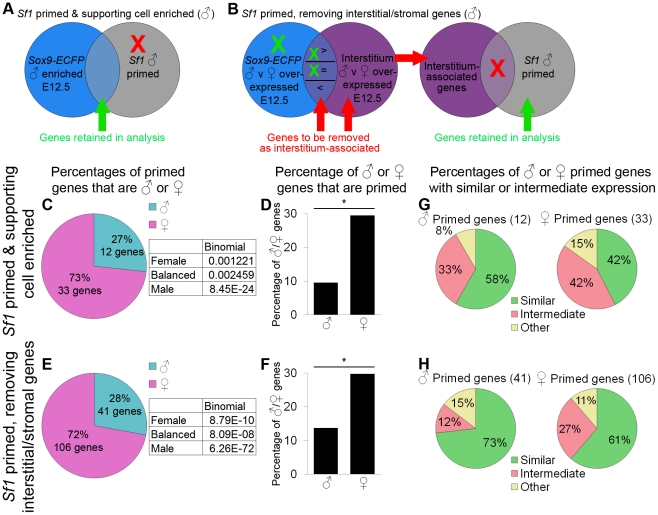
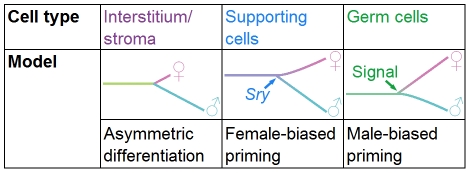
The divergence of distinct cell populations from multipotent progenitors is poorly understood, particularly in vivo. The gonad is an ideal place to study this process, because it originates as a bipotential primordium where multiple distinct lineages acquire sex-specific fates as the organ differentiates as a testis or an ovary. To gain a more detailed understanding of the process of gonadal differentiation at the level of the individual cell populations, we conducted microarrays on sorted cells from XX and XY mouse gonads at three time points spanning the period when the gonadal cells transition from sexually undifferentiated progenitors to their respective sex-specific fates. We analyzed supporting cells, interstitial/stromal cells, germ cells, and endothelial cells. This work identified genes specifically depleted and enriched in each lineage as it underwent sex-specific differentiation. We determined that the sexually undifferentiated germ cell and supporting cell progenitors showed lineage priming. We found that germ cell progenitors were primed with a bias toward the male fate. In contrast, supporting cells were primed with a female bias, indicative of the robust repression program involved in the commitment to XY supporting cell fate. This study provides a molecular explanation reconciling the female default and balanced models of sex determination and represents a rich resource for the field. More importantly, it yields new insights into the mechanisms by which different cell types in a single organ adopt their respective fates.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Koopman P, Gubbay J, Vivian N, Goodfellow P, Lovell-Badge R. Male development of chromosomally female mice transgenic for Sry. Nature. 1991;351:117–121. - PubMed
-
- Gubbay J, Collignon J, Koopman P, Capel B, Economou A, et al. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346:245–250. - PubMed
-
- Karl J, Capel B. Sertoli cells of the mouse testis originate from the coelomic epithelium. Dev Biol. 1998;203:323–333. - PubMed
-
- Wylie C. Germ cells. Cell. 1999;96:165–174. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
