

Evolution of the metabolic and regulatory networks associated with oxygen availability in two phytopathogenic enterobacteria
- PMID: 22439737
- PMCID: PMC3349551
- DOI: 10.1186/1471-2164-13-110
Evolution of the metabolic and regulatory networks associated with oxygen availability in two phytopathogenic enterobacteria
Abstract
Background: Dickeya dadantii and Pectobacterium atrosepticum are phytopathogenic enterobacteria capable of facultative anaerobic growth in a wide range of O2 concentrations found in plant and natural environments. The transcriptional response to O2 remains under-explored for these and other phytopathogenic enterobacteria although it has been well characterized for animal-associated genera including Escherichia coli and Salmonella enterica. Knowledge of the extent of conservation of the transcriptional response across orthologous genes in more distantly related species is useful to identify rates and patterns of regulon evolution. Evolutionary events such as loss and acquisition of genes by lateral transfer events along each evolutionary branch results in lineage-specific genes, some of which may have been subsequently incorporated into the O2-responsive stimulon. Here we present a comparison of transcriptional profiles measured using densely tiled oligonucleotide arrays for two phytopathogens, Dickeya dadantii 3937 and Pectobacterium atrosepticum SCRI1043, grown to mid-log phase in MOPS minimal medium (0.1% glucose) with and without O2.
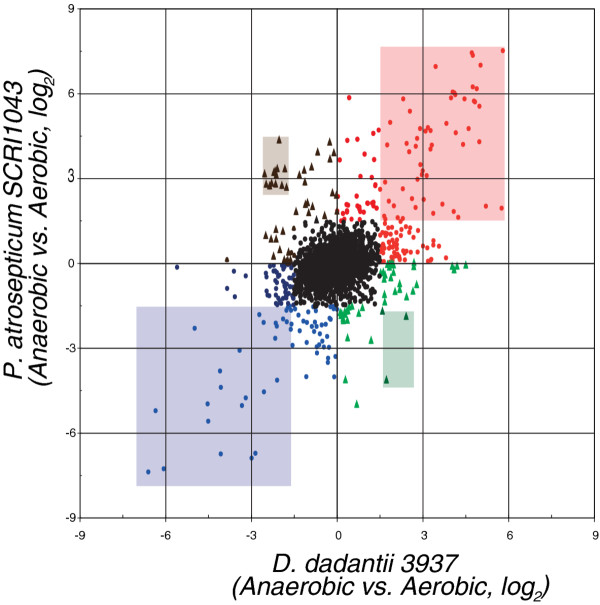
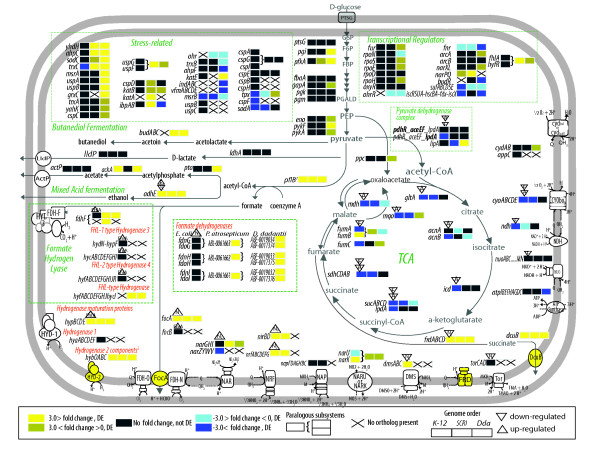
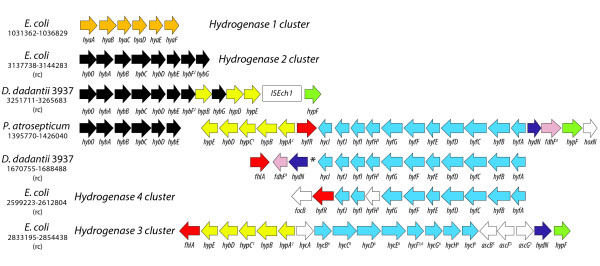
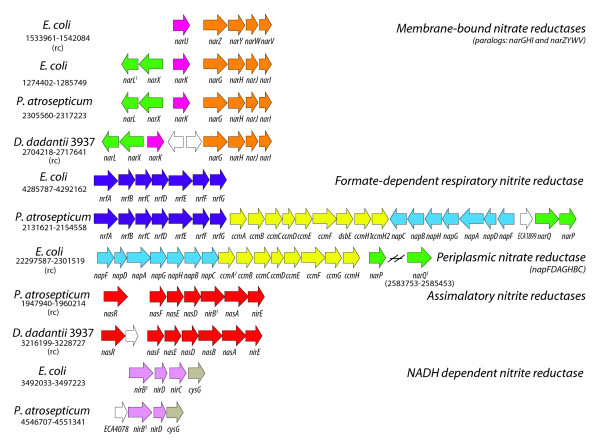
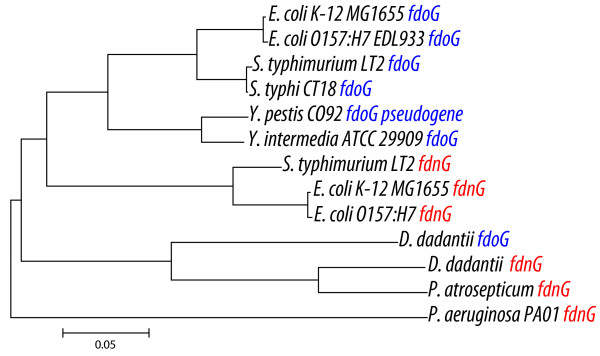
Results: More than 7% of the genes of each phytopathogen are differentially expressed with greater than 3-fold changes under anaerobic conditions. In addition to anaerobic metabolism genes, the O2 responsive stimulon includes a variety of virulence and pathogenicity-genes. Few of these genes overlap with orthologous genes in the anaerobic stimulon of E. coli. We define these as the conserved core, in which the transcriptional pattern as well as genetic architecture are well preserved. This conserved core includes previously described anaerobic metabolic pathways such as fermentation. Other components of the anaerobic stimulon show variation in genetic content, genome architecture and regulation. Notably formate metabolism, nitrate/nitrite metabolism, and fermentative butanediol production, differ between E. coli and the phytopathogens. Surprisingly, the overlap of the anaerobic stimulon between the phytopathogens is also relatively small considering that they are closely related, occupy similar niches and employ similar strategies to cause disease. There are cases of interesting divergences in the pattern of transcription of genes between Dickeya and Pectobacterium for virulence-associated subsystems including the type VI secretion system (T6SS), suggesting that fine-tuning of the stimulon impacts interaction with plants or competing microbes.
Conclusions: The small number of genes (an even smaller number if we consider operons) comprising the conserved core transcriptional response to O2 limitation demonstrates the extent of regulatory divergence prevalent in the Enterobacteriaceae. Our orthology-driven comparative transcriptomics approach indicates that the adaptive response in the eneterobacteria is a result of interaction of core (regulators) and lineage-specific (structural and regulatory) genes. Our subsystems based approach reveals that similar phenotypic outcomes are sometimes achieved by each organism using different genes and regulatory strategies.
Figures
References
-
- Perombelon MCM. Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol. 2002;51(1):1–12. 10.1046/j.0032-0862.2001.Short title.doc.x.
-
- Lemattre P. Chimie horticole, Nouvelle éd. edn. Paris,: J.-B. Baillière; 1972.
-
- Harrison MD, Nielsen LW. In: Compendium of Potato Diseases. Hooker WJ, St. Paul MN, editor. American Phytopathological Society; 1981. Blackleg and bacterial soft rot; pp. 27–29.
-
- Perombelon MCM, Kelman A. Ecology of the Soft Rot Erwinias. Annu Rev Phytopathol. 1980;18(1):361–387. doi: 10.1146/annurev.py.18.090180.002045. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
