

Mechanisms for recurrent and complex human genomic rearrangements
- PMID: 22440479
- PMCID: PMC3378805
- DOI: 10.1016/j.gde.2012.02.012
Mechanisms for recurrent and complex human genomic rearrangements
Abstract
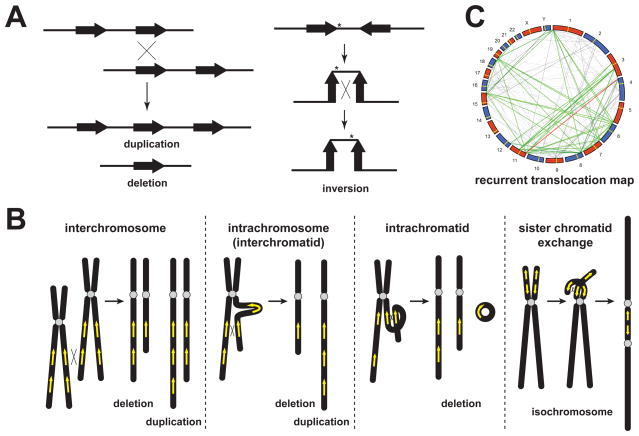
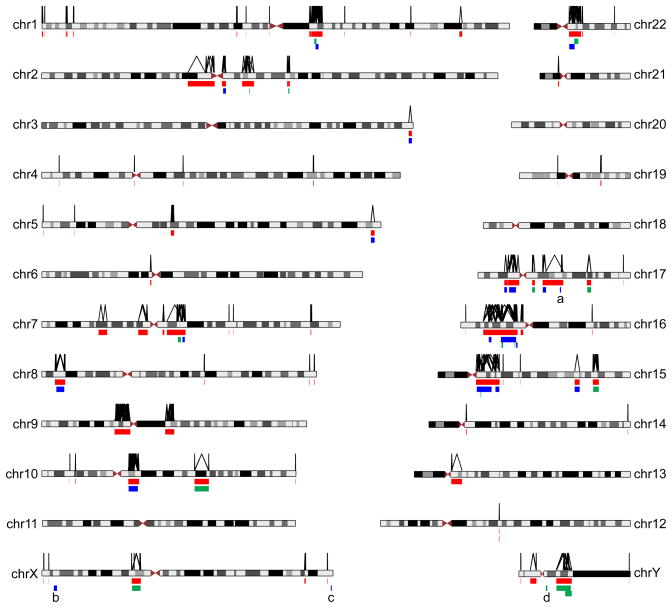
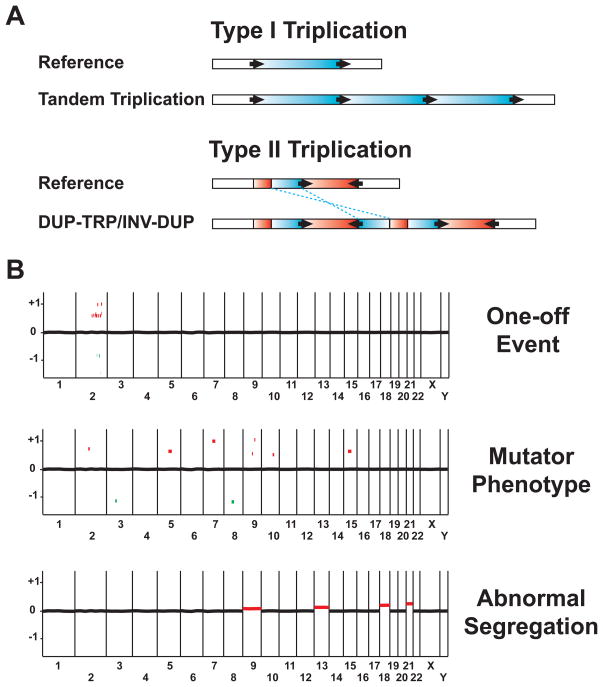
During the last two decades, the importance of human genome copy number variation (CNV) in disease has become widely recognized. However, much is not understood about underlying mechanisms. We show how, although model organism research guides molecular understanding, important insights are gained from study of the wealth of information available in the clinic. We describe progress in explaining nonallelic homologous recombination (NAHR), a major cause of copy number change occurring when control of allelic recombination fails, highlight the growing importance of replicative mechanisms to explain complex events, and describe progress in understanding extreme chromosome reorganization (chromothripsis). Both nonhomologous end-joining and aberrant replication have significant roles in chromothripsis. As we study CNV, the processes underlying human genome evolution are revealed.
Copyright © 2012 Elsevier Ltd. All rights reserved.
Figures
References
-
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. - PubMed
-
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. This study summarized structural variation findings in 185 individuals from the 1000 Genome Project pilot study. - PMC - PubMed
-
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, et al. Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. This study provided a comprehensive map of human genome CNV. They used high resolution array CGH to discover CNV, genotyped 4978 CNVs in different populations and assessed their functional impacts. - PMC - PubMed
-
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. Constructed a first-generation human genome CNV map from 270 HapMap individuals using SNP genotyping array and clone-based CGH. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
