

Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles
- PMID: 22442255
- PMCID: PMC3418848
- DOI: 10.1074/mcp.M111.015362
Super-SILAC allows classification of diffuse large B-cell lymphoma subtypes by their protein expression profiles
Abstract
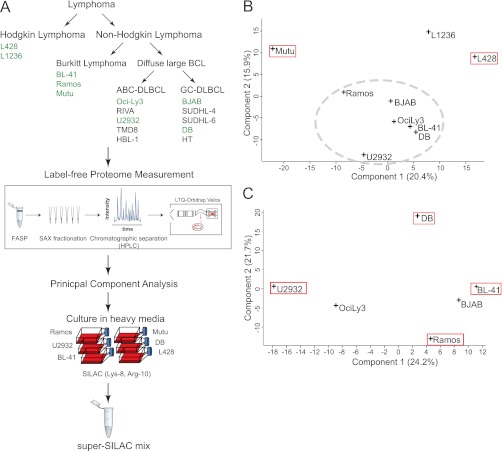
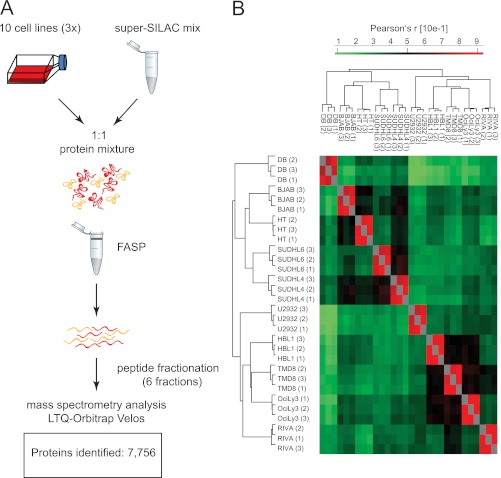
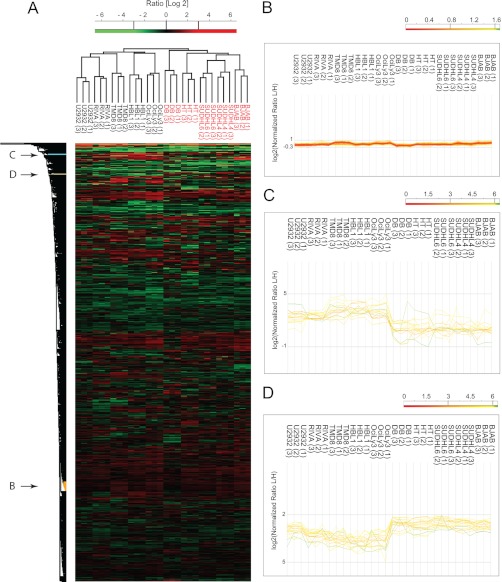
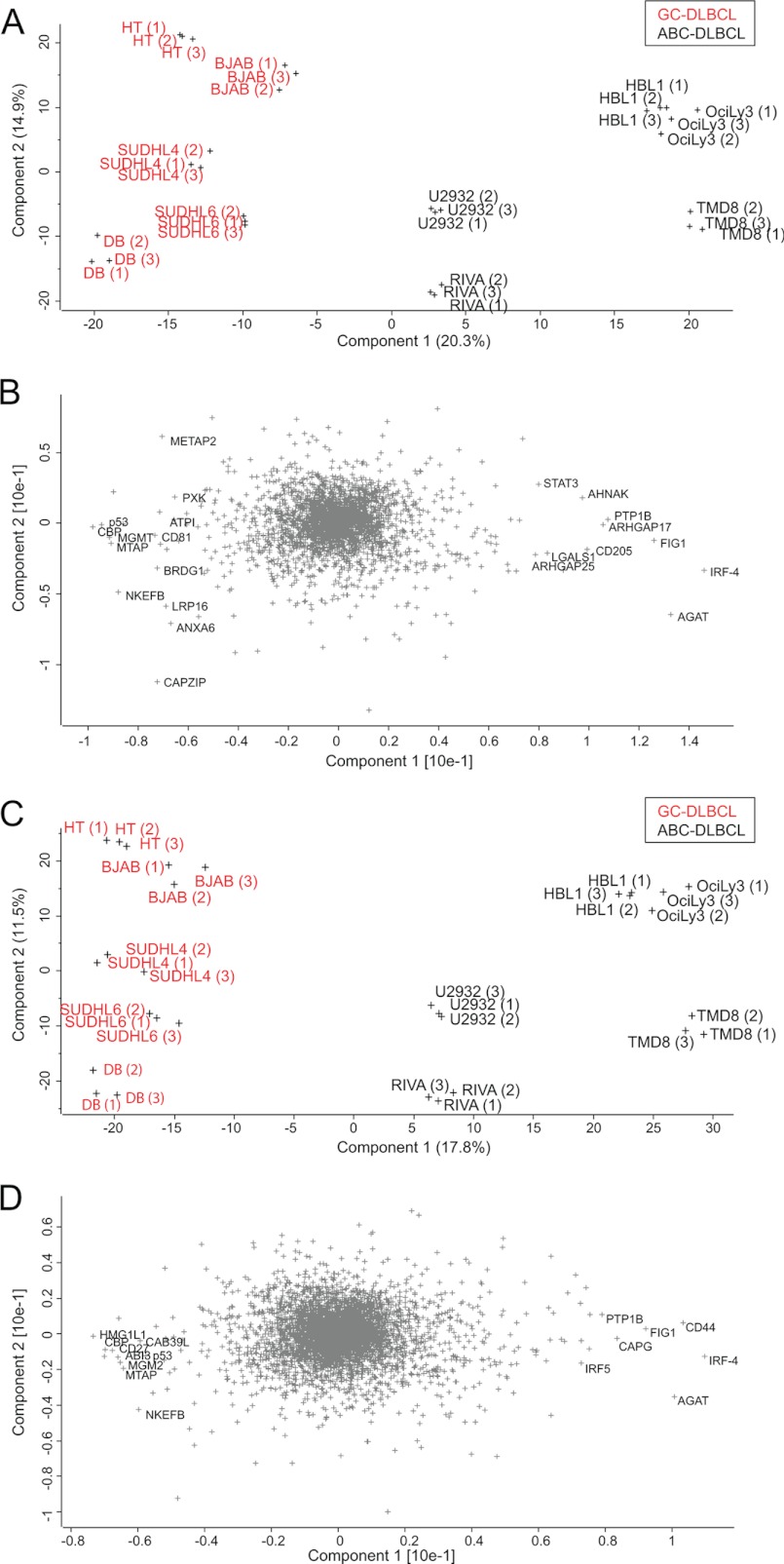
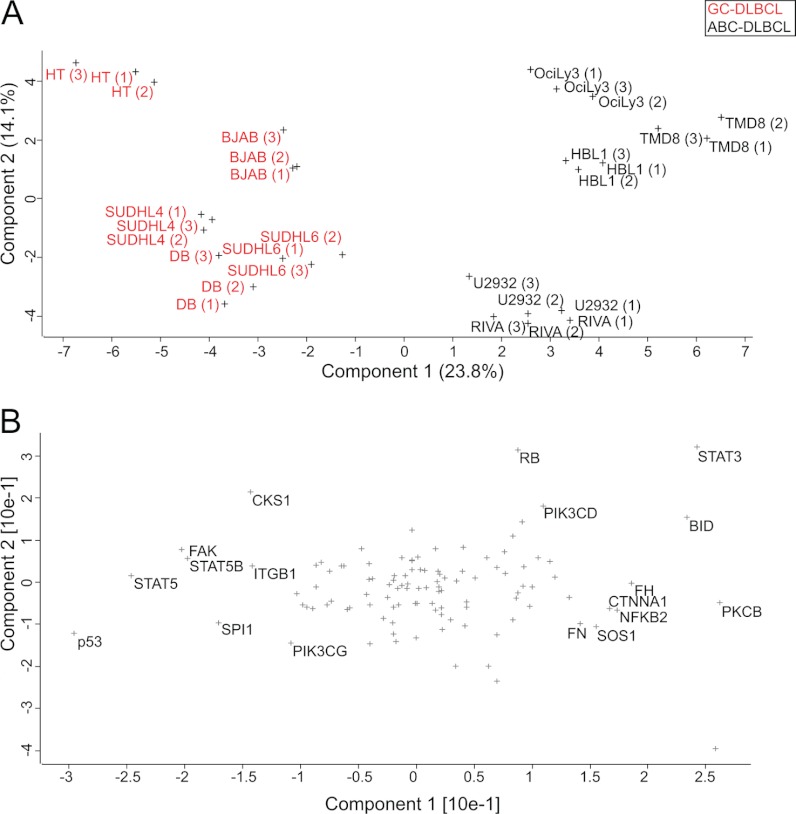
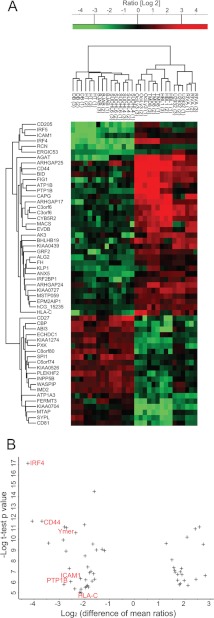
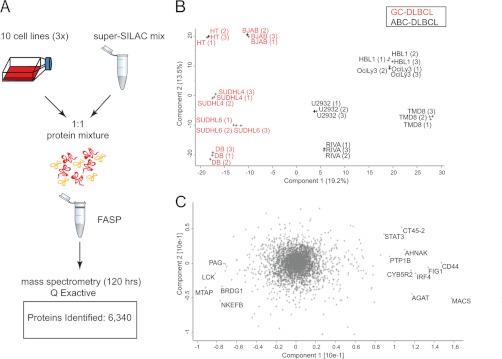
Correct classification of cancer patients into subtypes is a prerequisite for acute diagnosis and effective treatment. Currently this classification relies mainly on histological assessment, but gene expression analysis by microarrays has shown great promise. Here we show that high accuracy, quantitative proteomics can robustly segregate cancer subtypes directly at the level of expressed proteins. We investigated two histologically indistinguishable subtypes of diffuse large B-cell lymphoma (DLBCL): activated B-cell-like (ABC) and germinal-center B-cell-like (GCB) subtypes, by first developing a general lymphoma stable isotope labeling with amino acids in cell culture (SILAC) mix from heavy stable isotope-labeled cell lines. This super-SILAC mix was combined with cell lysates from five ABC-DLBCL and five GCB-DLBCL cell lines. Shotgun proteomic analysis on a linear ion trap Orbitrap mass spectrometer with high mass accuracy at the MS and MS/MS levels yielded a proteome of more than 7,500 identified proteins. High accuracy of quantification allowed robust separation of subtypes by principal component analysis. The main contributors to the classification included proteins known to be differentially expressed between the subtypes such as the transcription factors IRF4 and SPI1/PU.1, cell surface markers CD44 and CD27, as well as novel candidates. We extracted a signature of 55 proteins that segregated subtypes and contained proteins connected to functional differences between the ABC and GCB-DLBCL subtypes, including many NF-κB-regulated genes. Shortening the analysis time to single-shot analysis combined with use of the new linear quadrupole Orbitrap analyzer (Q Exactive) also clearly differentiated between the subtypes. These results show that high resolution shotgun proteomics combined with super-SILAC-based quantification is a promising new technology for tumor characterization and classification.
Figures
References
-
- Quackenbush J. (2006) Microarray Analysis and Tumor Classification. New Engl. J. Med. 354, 2463–2472 - PubMed
-
- McDermott U., Downing J. R., Stratton M. R. (2011) Genomics and the Continuum of Cancer Care. New Engl. J. Med. 364, 340–350 - PubMed
-
- Hanash S., Taguchi A. (2010) The grand challenge to decipher the cancer proteome. Nat. Rev. Cancer 10, 652–660 - PubMed
-
- Choudhary C., Mann M. (2010) Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell Biol. 11, 427–439 - PubMed
-
- Aebersold R., Mann M. (2003) Mass spectrometry-based proteomics. Nature 422, 198–207 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
