

Endoplasmic reticulum stress and type 2 diabetes
- PMID: 22443930
- PMCID: PMC3684428
- DOI: 10.1146/annurev-biochem-072909-095555
Endoplasmic reticulum stress and type 2 diabetes
Abstract
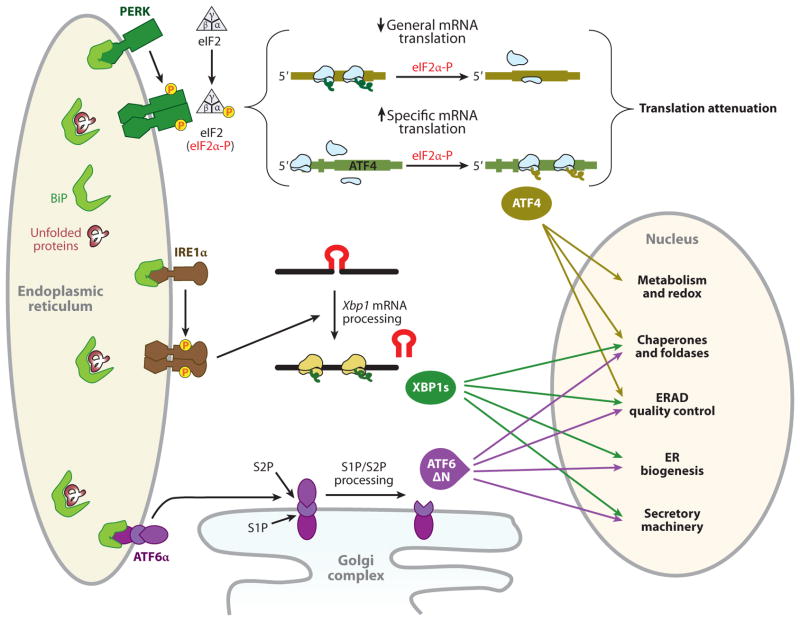
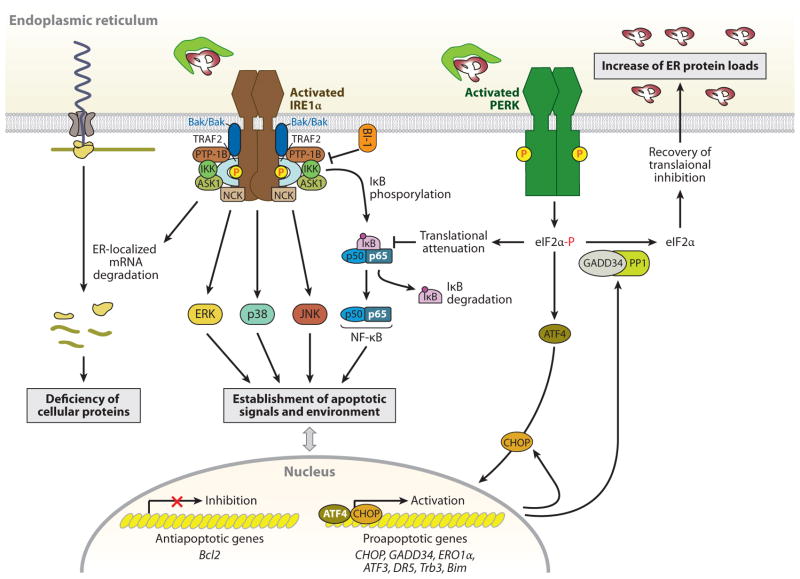
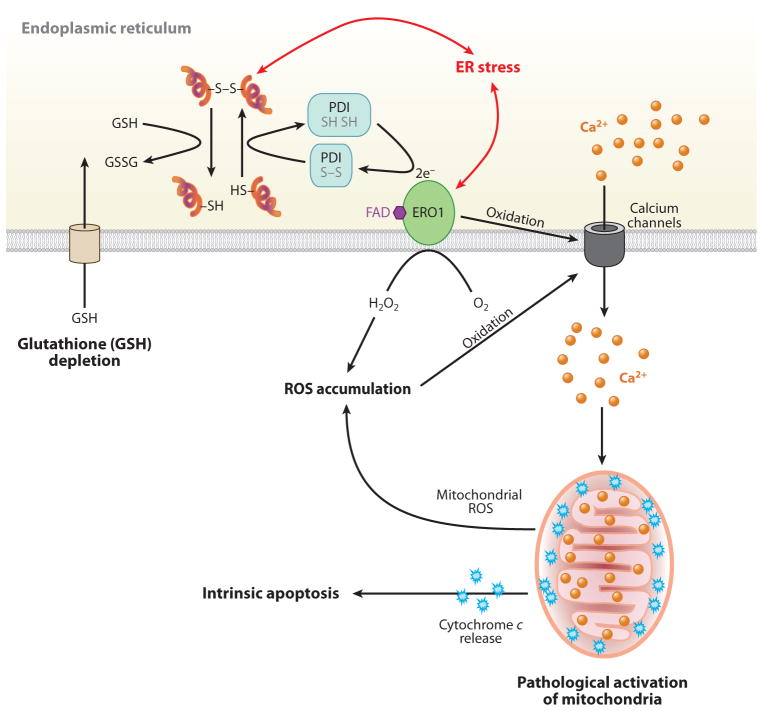
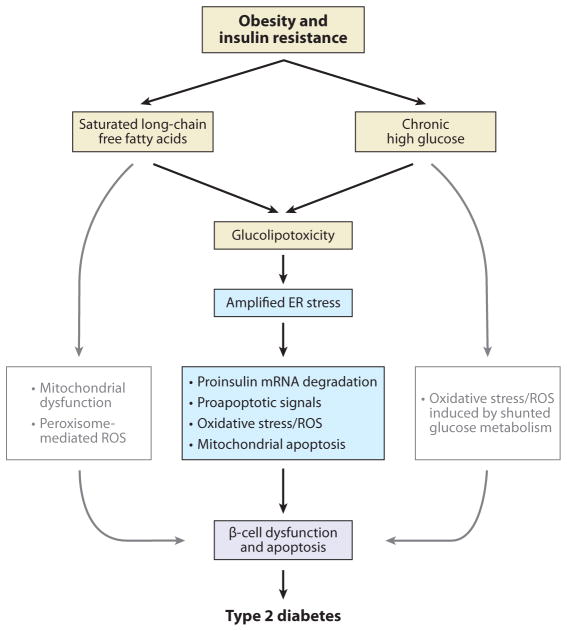
Given the functional importance of the endoplasmic reticulum (ER), an organelle that performs folding, modification, and trafficking of secretory and membrane proteins to the Golgi compartment, the maintenance of ER homeostasis in insulin-secreting β-cells is very important. When ER homeostasis is disrupted, the ER generates adaptive signaling pathways, called the unfolded protein response (UPR), to maintain homeostasis of this organelle. However, if homeostasis fails to be restored, the ER initiates death signaling pathways. New observations suggest that both chronic hyperglycemia and hyperlipidemia, known as important causative factors of type 2 diabetes (T2D), disrupt ER homeostasis to induce unresolvable UPR activation and β-cell death. This review examines how the UPR pathways, induced by high glucose and free fatty acids (FFAs), interact to disrupt ER function and cause β-cell dysfunction and death.
Figures

References
-
- Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract. 2010;87:4–14. - PubMed
-
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–46. - PubMed
-
- Jetton TL, Lausier J, LaRock K, Trotman WE, Larmie B, et al. Mechanisms of compensatory beta-cell growth in insulin-resistant rats: roles of Akt kinase. Diabetes. 2005;54:2294–304. - PubMed
-
- Liu YQ, Jetton TL, Leahy JL. β-cell adaptation to insulin resistance. Increased pyruvate carboxylase and malate-pyruvate shuttle activity in islets of nondiabetic Zucker fatty rats. J Biol Chem. 2002;277:39163–68. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
