

Macrophage autophagy plays a protective role in advanced atherosclerosis
- PMID: 22445600
- PMCID: PMC3322248
- DOI: 10.1016/j.cmet.2012.01.022
Macrophage autophagy plays a protective role in advanced atherosclerosis
Abstract
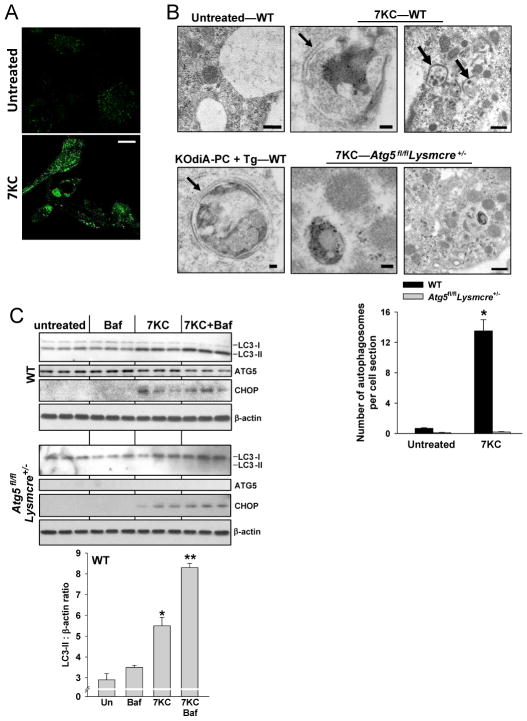
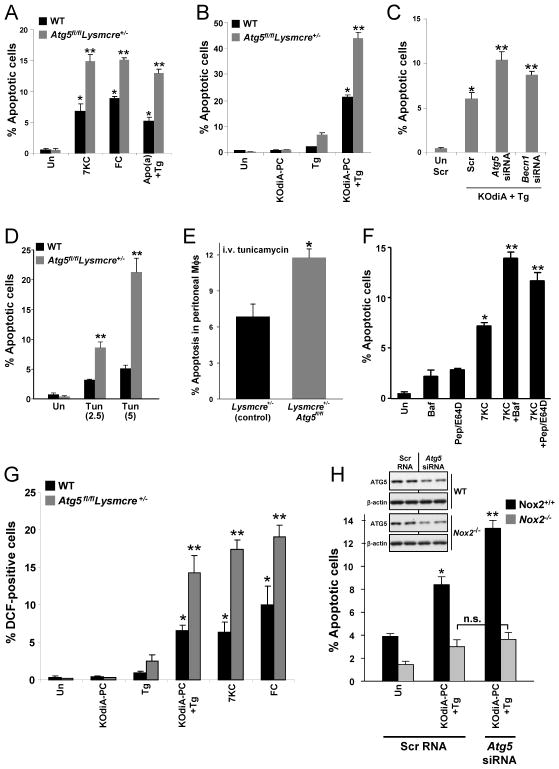
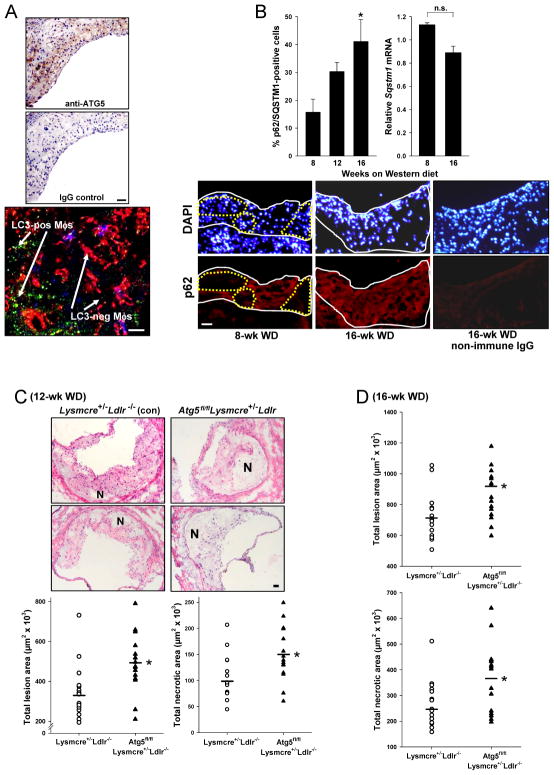
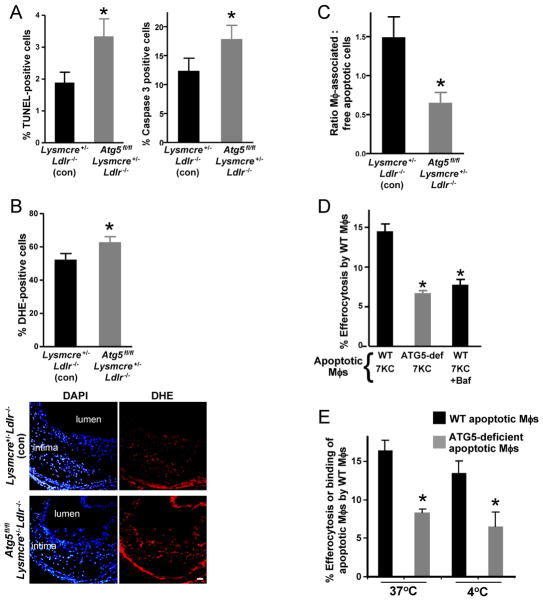
In advanced atherosclerosis, macrophage apoptosis coupled with defective phagocytic clearance of the apoptotic cells (efferocytosis) promotes plaque necrosis, which precipitates acute atherothrombotic cardiovascular events. Oxidative and endoplasmic reticulum (ER) stress in macrophages are important causes of advanced lesional macrophage apoptosis. We now show that proapoptotic oxidative/ER stress inducers trigger another stress reaction in macrophages, autophagy. Inhibition of autophagy by silencing ATG5 or other autophagy mediators enhances apoptosis and NADPH oxidase-mediated oxidative stress while at the same time rendering the apoptotic cells less well recognized by efferocytes. Most importantly, macrophage ATG5 deficiency in fat-fed Ldlr(-/-) mice increases apoptosis and oxidative stress in advanced lesional macrophages, promotes plaque necrosis, and worsens lesional efferocytosis. These findings reveal a protective process in oxidatively stressed macrophages relevant to plaque necrosis, suggesting a mechanism-based strategy to therapeutically suppress atherosclerosis progression and its clinical sequelae.
Copyright © 2012 Elsevier Inc. All rights reserved.
Figures
Comment in
-
How to chew up cells: lessons for the atherosclerotic plaque.Circ Res. 2012 Aug 31;111(6):669-71. doi: 10.1161/CIRCRESAHA.112.268151. Circ Res. 2012. PMID: 22935531 No abstract available.
References
-
- Ball RY, Stowers EC, Burton JH, Cary NR, Skepper JN, Mitchinson MJ. Evidence that the death of macrophage foam cells contributes to the lipid core of atheroma. Atherosclerosis. 1995;114:45–54. - PubMed
-
- Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181–197. - PubMed
-
- Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de FU, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. - PubMed
-
- Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. - PubMed
-
- De Meyer GR, Martinet W. Autophagy in the cardiovascular system. Biochim Biophys Acta. 2009;1793:1485–1495. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
