

Genetic variants influencing human aging from late-onset Alzheimer's disease (LOAD) genome-wide association studies (GWAS)
- PMID: 22445811
- PMCID: PMC4120742
- DOI: 10.1016/j.neurobiolaging.2012.02.014
Genetic variants influencing human aging from late-onset Alzheimer's disease (LOAD) genome-wide association studies (GWAS)
Abstract
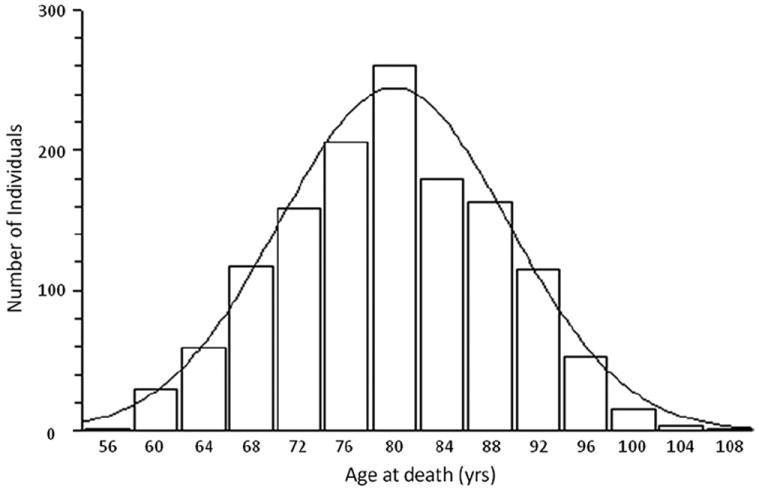
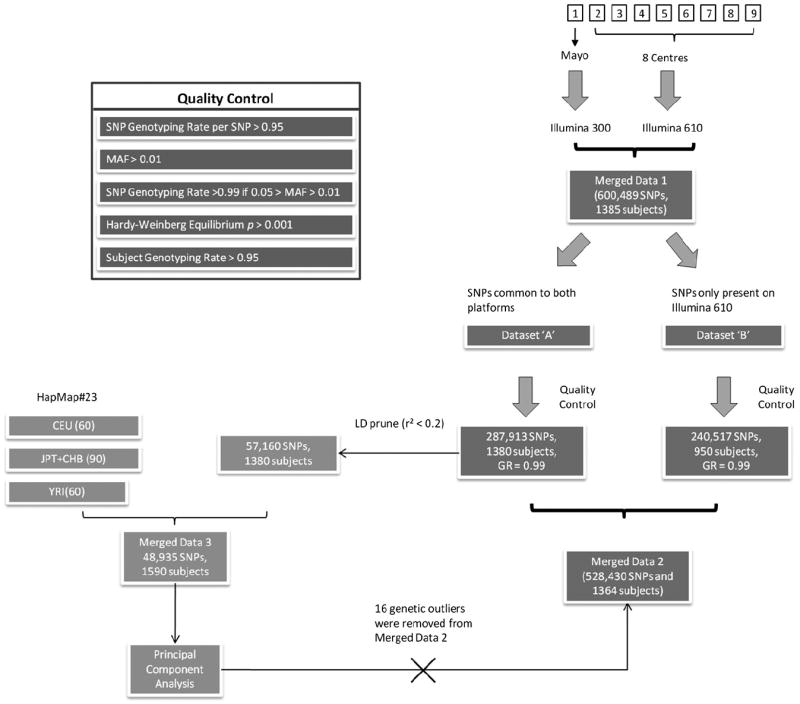
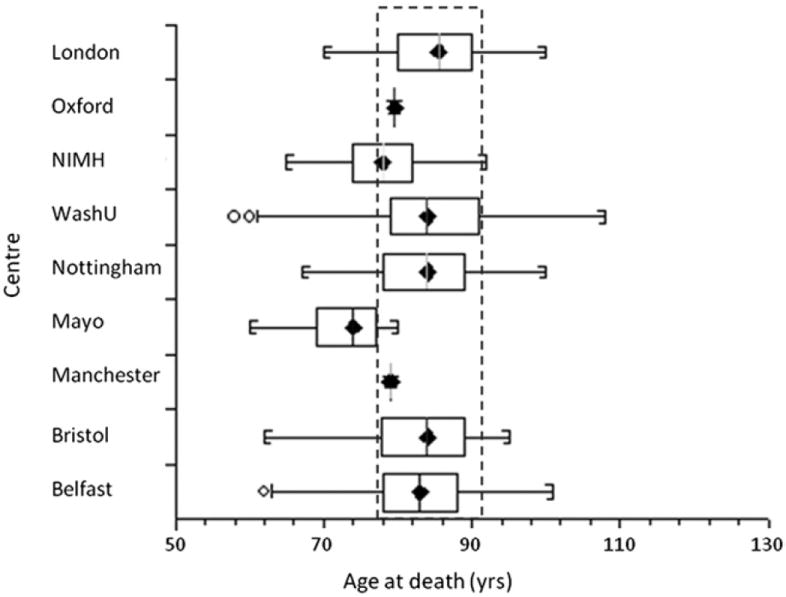
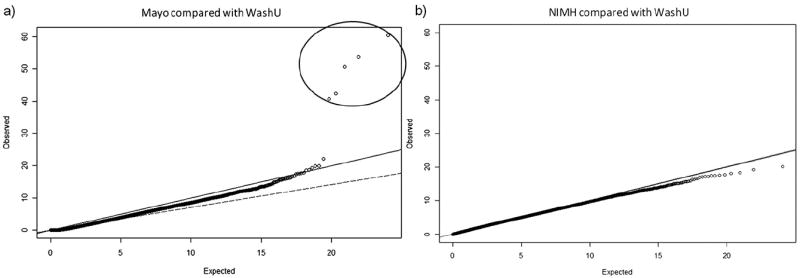
Genetics plays a crucial role in human aging with up to 30% of those living to the mid-80s being determined by genetic variation. Survival to older ages likely entails an even greater genetic contribution. There is increasing evidence that genes implicated in age-related diseases, such as cancer and neuronal disease, play a role in affecting human life span. We have selected the 10 most promising late-onset Alzheimer's disease (LOAD) susceptibility genes identified through several recent large genome-wide association studies (GWAS). These 10 LOAD genes (APOE, CLU, PICALM, CR1, BIN1, ABCA7, MS4A6A, CD33, CD2AP, and EPHA1) have been tested for association with human aging in our dataset (1385 samples with documented age at death [AAD], age range: 58-108 years; mean age at death: 80.2) using the most significant single nucleotide polymorphisms (SNPs) found in the previous studies. Apart from the APOE locus (rs2075650) which showed compelling evidence of association with risk on human life span (p = 5.27 × 10(-4)), none of the other LOAD gene loci demonstrated significant evidence of association. In addition to examining the known LOAD genes, we carried out analyses using age at death as a quantitative trait. No genome-wide significant SNPs were discovered. Increasing sample size and statistical power will be imperative to detect genuine aging-associated variants in the future. In this report, we also discuss issues relating to the analysis of genome-wide association studies data from different centers and the bioinformatic approach required to distinguish spurious genome-wide significant signals from real SNP associations.
Copyright © 2012 Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Approval was obtained from the ethics committee or institutional review board of each institution responsible for the ascertainment and collection of samples. Written informed consent was obtained for all individuals who participated in this study.
Figures
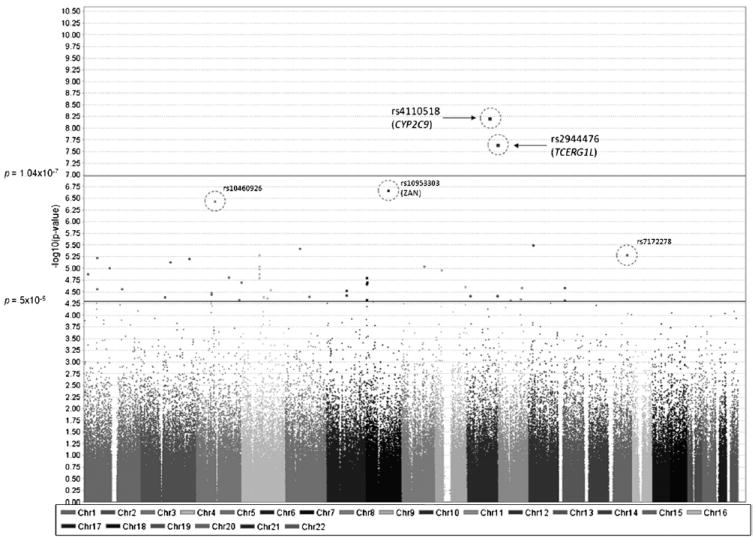
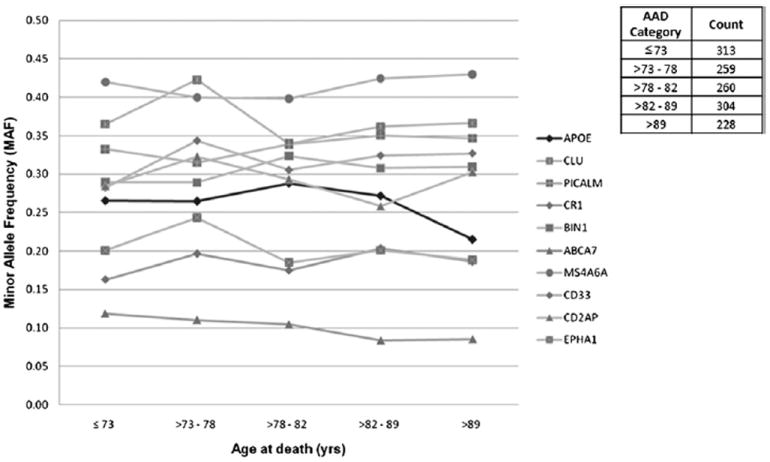
References
-
- Arking DE, Atzmon G, Arking A, Barzilai N, Dietz HC. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ Res. 2005;96:412–418. - PubMed
-
- Atzmon G, Schechter C, Greiner W, Davidson D, Rennert G, Barzilai N. Clinical phenotype of families with longevity. J Am Geriatr Soc. 2004;52:274–277. - PubMed
-
- Aulchenko YS, Ripke S, Isaacs A, van Duijn CM. GenABEL: an R library for genome-wide association analysis. Bioinformatics. 2007;23:1294–1296. - PubMed
-
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. - PubMed
-
- Barzilai N, Atzmon G, Schechter C, Schaefer EJ, Cupples AL, Lipton R, Cheng S, Shuldiner AR. Unique lipoprotein phenotype and genotype associated with exceptional longevity. Jama. 2003;290:2030–2040. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
