

Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma
- PMID: 22447568
- PMCID: PMC3319869
- DOI: 10.1158/0008-5472.CAN-11-3831
Hypoxia-induced autophagy promotes tumor cell survival and adaptation to antiangiogenic treatment in glioblastoma
Abstract
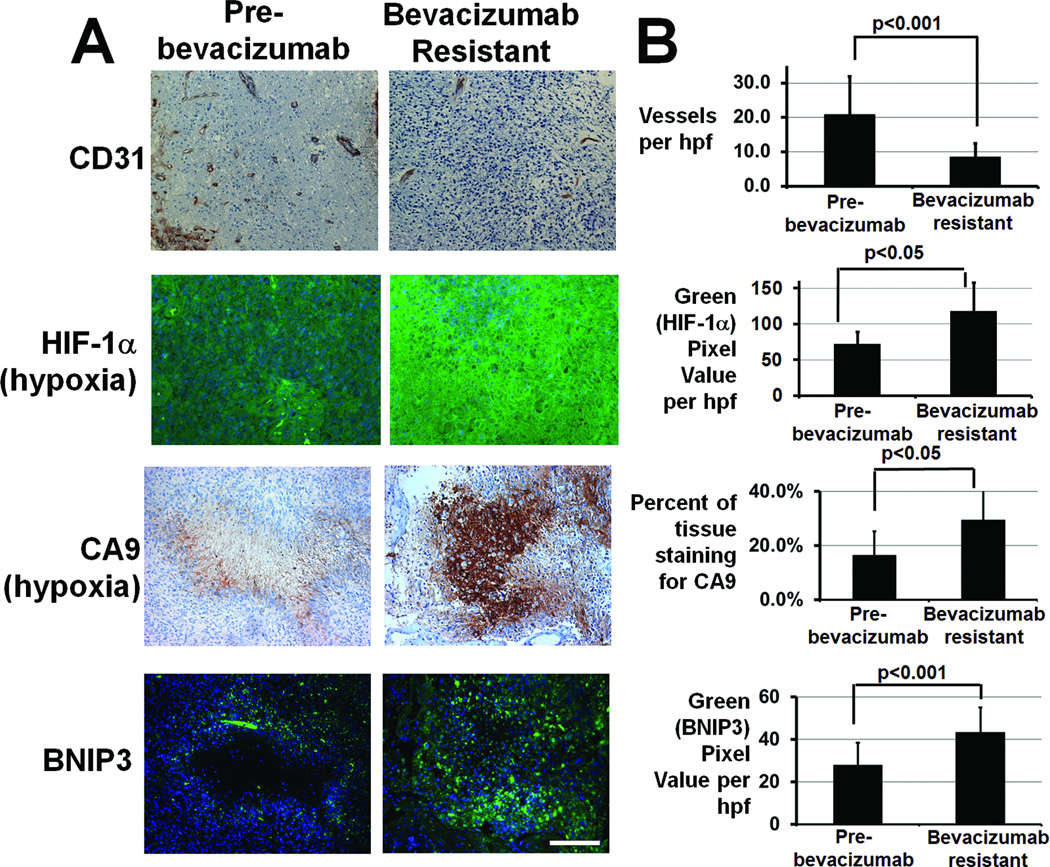
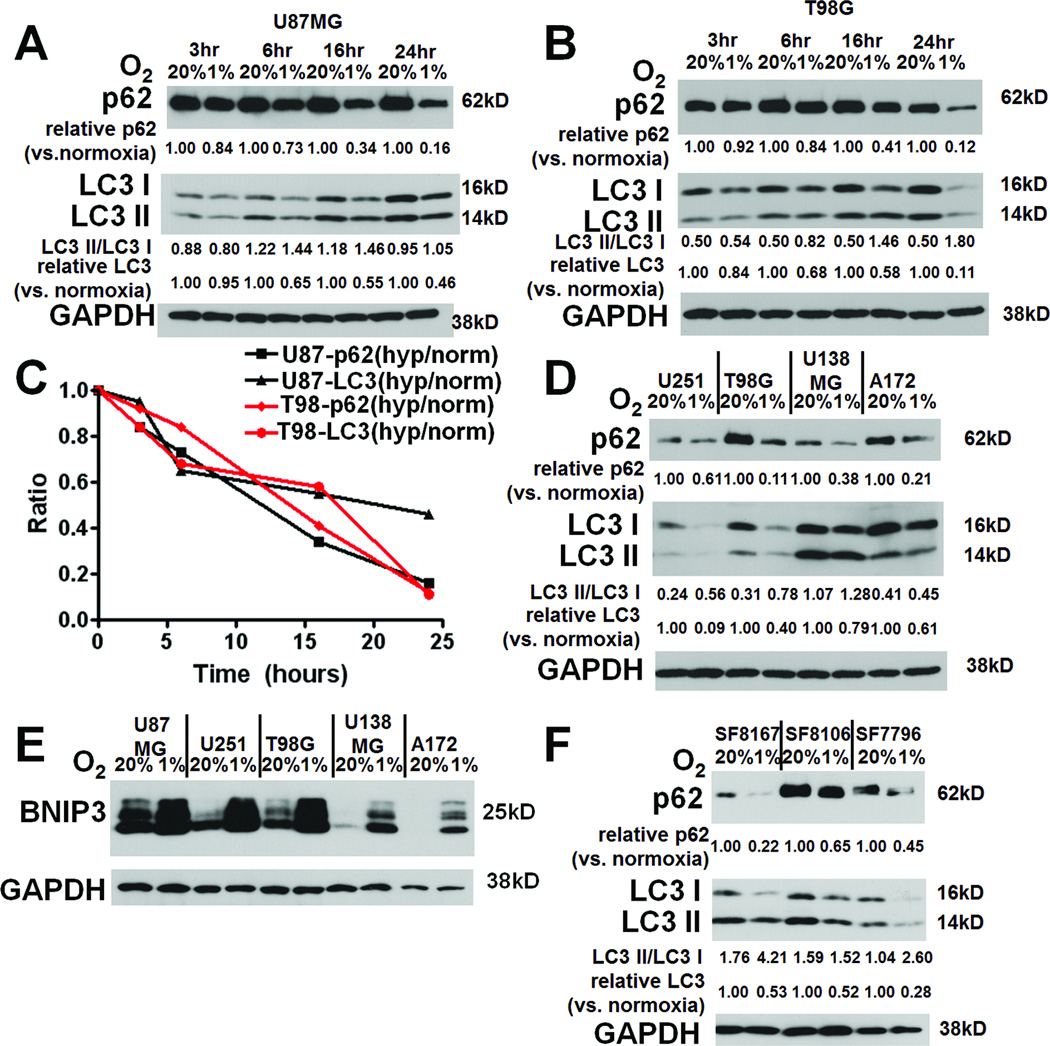
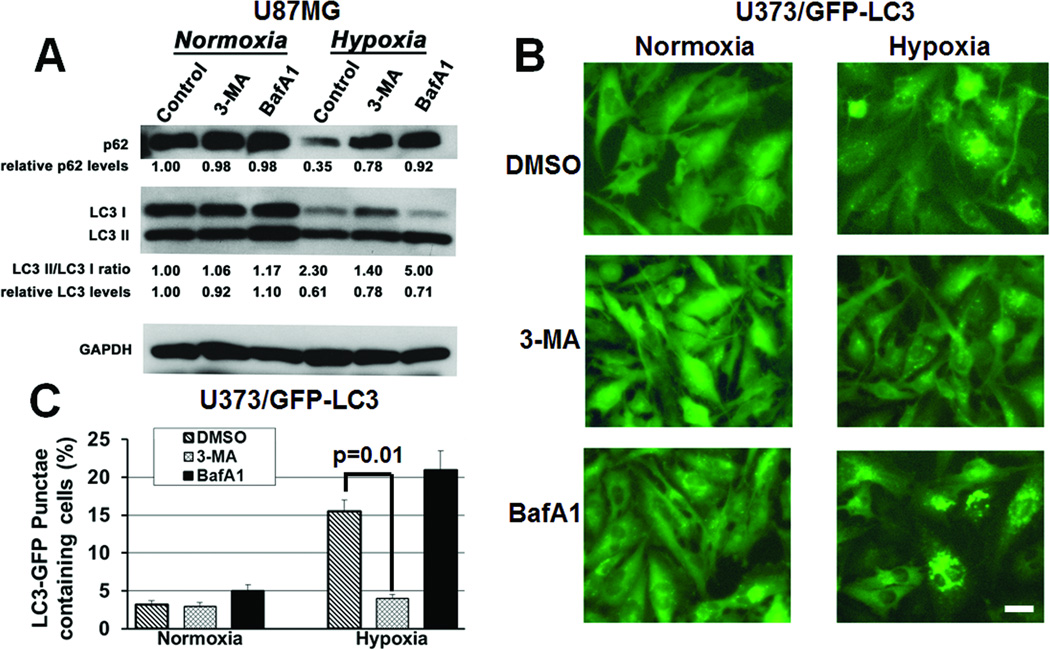
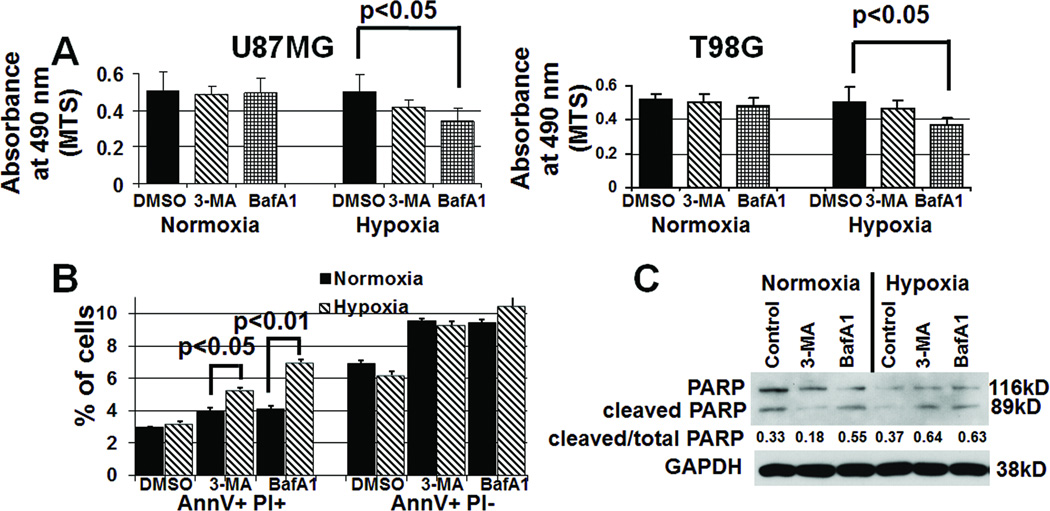
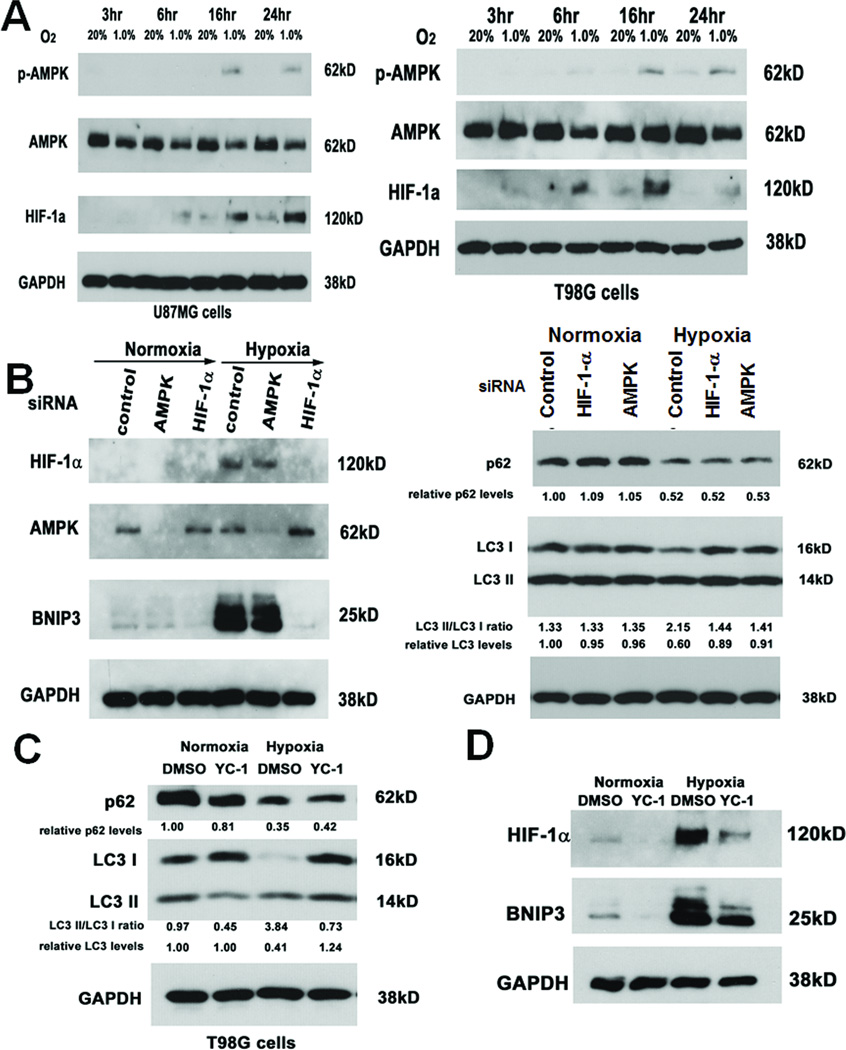
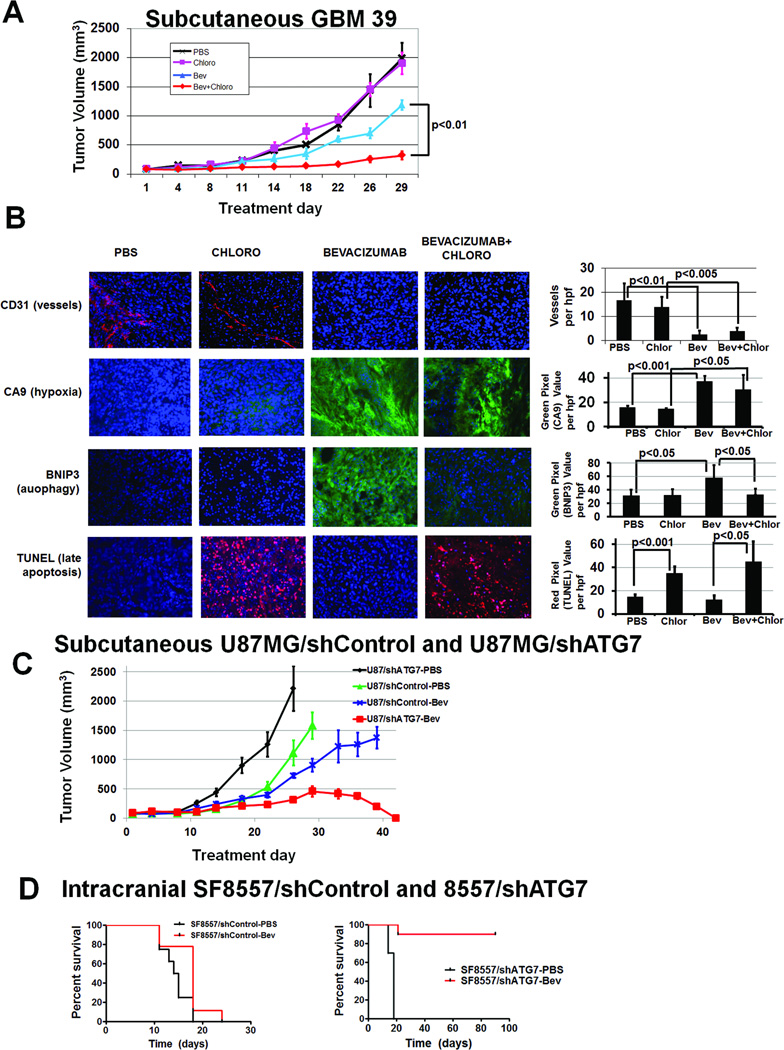
Antiangiogenic therapy leads to devascularization that limits tumor growth. However, the benefits of angiogenesis inhibitors are typically transient and resistance often develops. In this study, we explored the hypothesis that hypoxia caused by antiangiogenic therapy induces tumor cell autophagy as a cytoprotective adaptive response, thereby promoting treatment resistance. Hypoxia-induced autophagy was dependent on signaling through the hypoxia-inducible factor-1α (HIF-1α)/AMPK pathway, and treatment of hypoxic cells with autophagy inhibitors caused a shift from autophagic to apoptotic cell death in vitro. In glioblastomas, clinically resistant to the VEGF-neutralizing antibody bevacizumab, increased regions of hypoxia and higher levels of autophagy-mediating BNIP3 were found when compared with pretreatment specimens from the same patients. When treated with bevacizumab alone, human glioblastoma xenografts showed increased BNIP3 expression and hypoxia-associated growth, which could be prevented by addition of the autophagy inhibitor chloroquine. In vivo targeting of the essential autophagy gene ATG7 also disrupted tumor growth when combined with bevacizumab treatment. Together, our findings elucidate a novel mechanism of resistance to antiangiogenic therapy in which hypoxia-mediated autophagy promotes tumor cell survival. One strong implication of our findings is that autophagy inhibitors may help prevent resistance to antiangiogenic therapy used in the clinic.
©2012 AACR.
Conflict of interest statement
No conflicts of interest to report.
Figures
References
-
- Jain RK. Lessons from multidisciplinary translational trials on anti-angiogenic therapy of cancer. Nat Rev Cancer. 2008;8:309–316. - PubMed
-
- Vredenburgh JJ, Desjardins A, Herndon JE, 2nd, Dowell JM, Reardon DA, Quinn JA, et al. Phase II trial of bevacizumab and irinotecan in recurrent malignant glioma. Clin Cancer Res. 2007;13:1253–1259. - PubMed
-
- Clark AJ, Lamborn KR, Butowski NA, Chang SM, Prados MD, Clarke JL, et al. Neurosurgical management and prognosis of patients with glioblastoma that progress during bevacizumab treatment. Neurosurgery. 2011 - PubMed
-
- Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22:177–180. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
