

Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans
- PMID: 22448145
- PMCID: PMC3308940
- DOI: 10.1371/journal.pbio.1001288
Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans
Abstract
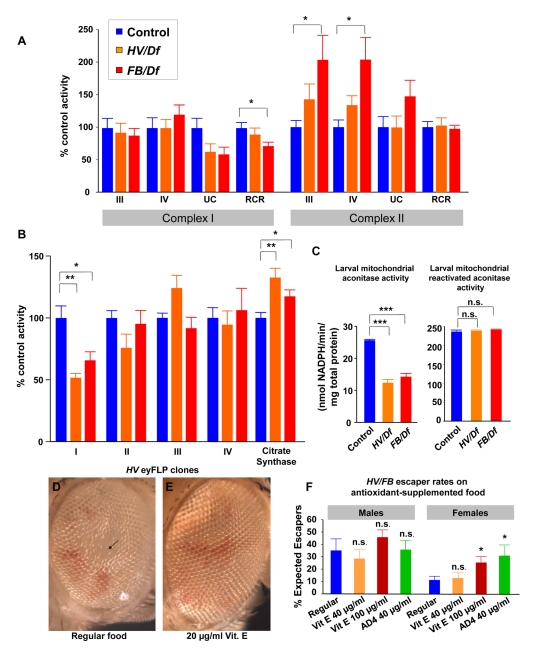
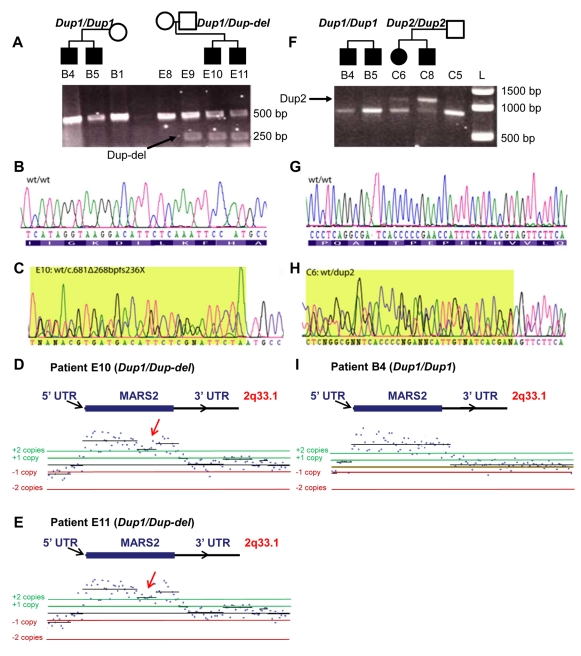
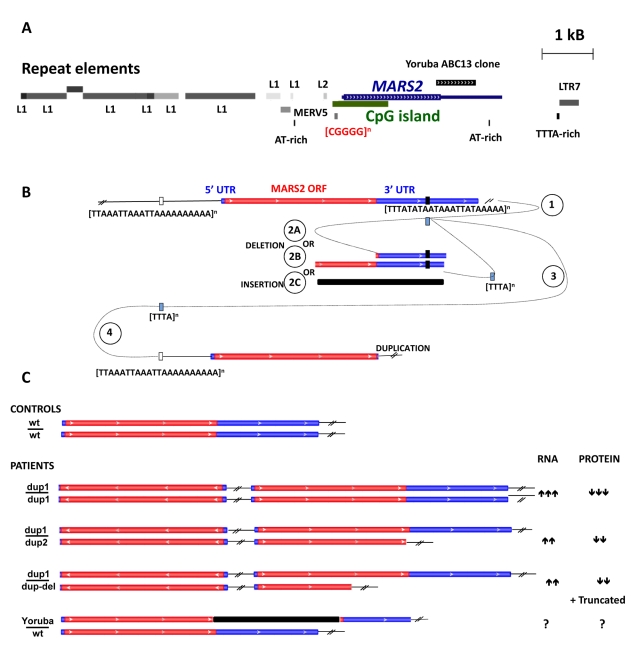
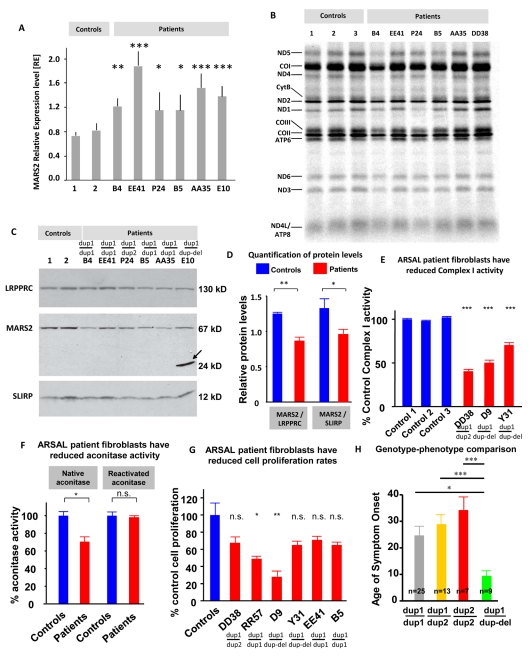
An increasing number of genes required for mitochondrial biogenesis, dynamics, or function have been found to be mutated in metabolic disorders and neurological diseases such as Leigh Syndrome. In a forward genetic screen to identify genes required for neuronal function and survival in Drosophila photoreceptor neurons, we have identified mutations in the mitochondrial methionyl-tRNA synthetase, Aats-met, the homologue of human MARS2. The fly mutants exhibit age-dependent degeneration of photoreceptors, shortened lifespan, and reduced cell proliferation in epithelial tissues. We further observed that these mutants display defects in oxidative phosphorylation, increased Reactive Oxygen Species (ROS), and an upregulated mitochondrial Unfolded Protein Response. With the aid of this knowledge, we identified MARS2 to be mutated in Autosomal Recessive Spastic Ataxia with Leukoencephalopathy (ARSAL) patients. We uncovered complex rearrangements in the MARS2 gene in all ARSAL patients. Analysis of patient cells revealed decreased levels of MARS2 protein and a reduced rate of mitochondrial protein synthesis. Patient cells also exhibited reduced Complex I activity, increased ROS, and a slower cell proliferation rate, similar to Drosophila Aats-met mutants.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
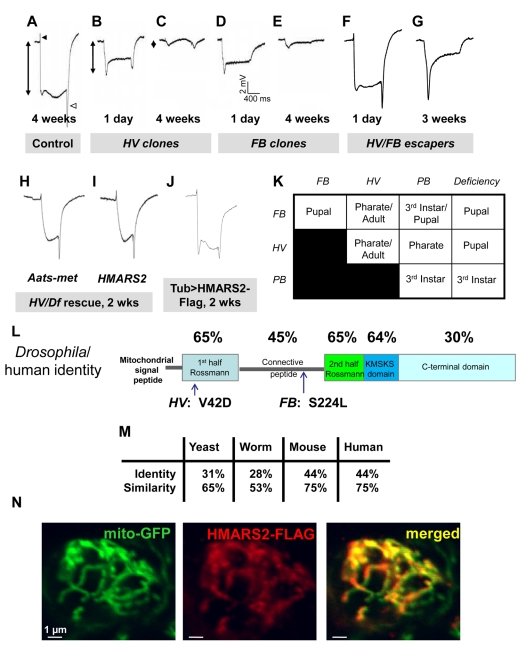
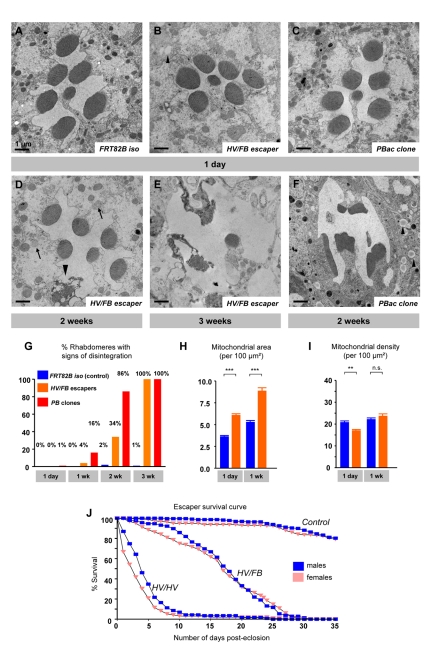
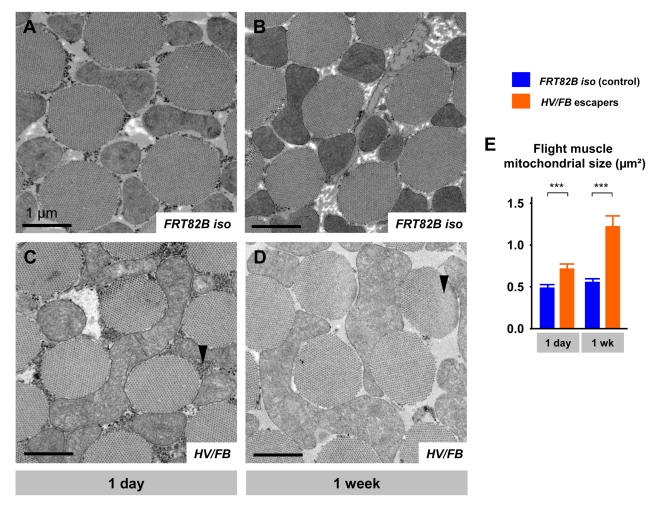
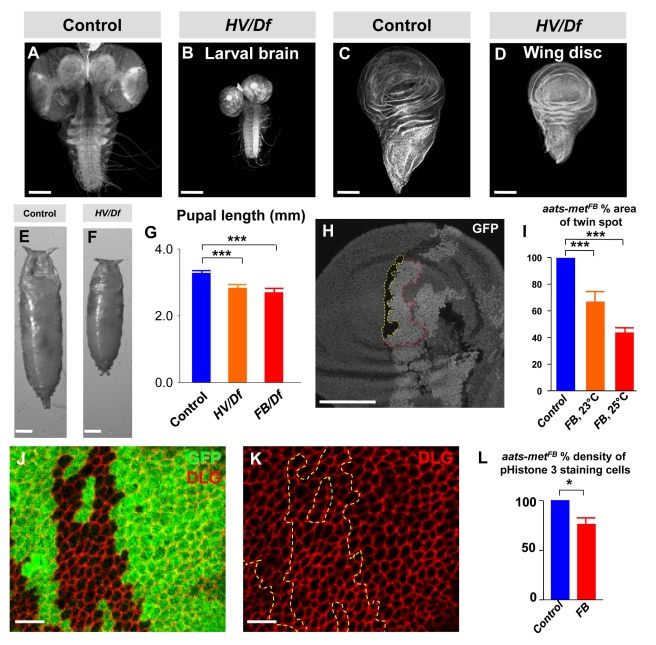
References
-
- Schapira A. H. Mitochondrial disease. Lancet. 2006;368:70–82. - PubMed
-
- Wallace D. C. Mitochondrial DNA mutations in disease and aging. Environmental and Molecular Mutagenesis. 2010;51:440–450. - PubMed
-
- Berger I, Hershkovitz E, Shaag A, Edvardson S, Saada A, et al. Mitochondrial complex I deficiency caused by a deleterious NDUFA11 mutation. Ann Neurol. 2008;63:405–408. - PubMed
-
- Hudson G, Amati-Bonneau P, Blakely E. L, Stewart J. D, He L, et al. Mutation of OPA1 causes dominant optic atrophy with external ophthalmoplegia, ataxia, deafness and multiple mitochondrial DNA deletions: a novel disorder of mtDNA maintenance. Brain. 2008;131:329–337. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
