

Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes
- PMID: 22450714
- PMCID: PMC3774053
- DOI: 10.1007/s10545-012-9477-y
Diversity of approaches to classic galactosemia around the world: a comparison of diagnosis, intervention, and outcomes
Erratum in
- J Inherit Metab Dis. 2012 Nov;35(6):1157. Scheweitzer-Krantz, Susanne [corrected to Schweitzer-Krantz, Susanne]
Abstract
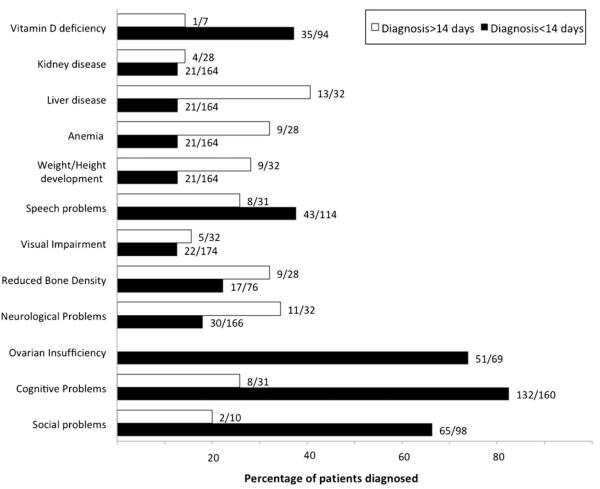
Without intervention, classic galactosemia is a potentially fatal disorder in infancy. With the benefit of early diagnosis and dietary restriction of galactose, the acute sequelae of classic galactosemia can be prevented or reversed. However, despite early and lifelong dietary treatment, many galactosemic patients go on to experience serious long-term complications including cognitive disability, speech problems, neurological and/or movement disorders and, in girls and women, ovarian dysfunction. Further, there remains uncertainty surrounding what constitutes a 'best practice' for treating this disorder. To explore the extent and implications of this uncertainty, we conducted a small but global survey of healthcare providers who follow patients with classic galactosemia, seeking to compare established protocols for diagnosis, intervention, and follow-up, as well as the outcomes and outcome frequencies seen in the patient populations cared for by these providers. We received 13 survey responses representing five continents and 11 countries. Respondents underscored disparities in approaches to diagnosis, management and follow-up care. Notably, we saw no clear relationship between differing approaches to care and long-term outcomes in the populations studied. Negative outcomes occurred in the majority of cases regardless of when treatment was initiated, how tightly galactose intake was restricted, or how closely patients were monitored. We document here what is, to our knowledge, the first global comparison of healthcare approaches to classic galactosemia. These data reinforce the idea that there is currently no one best practice for treating patients with classic galactosemia, and underscore the need for more extensive and statistically powerful comparative studies to reveal potential positive or negative impacts of differing approaches.
Figures
References
-
- Anasti J, Kalantaridou S, Kimzey L, Defensor R, Nelson L. Bone loss in young women with karyotypically normal spontaneous premature ovarian failure. Obstet Gynecol. 1998;91:12–15. - PubMed
-
- Antshel K, Epstein I, Waisbren S. Cognitive strengths and weaknesses in children and adolescents homozygous for the galactosemia Q188R mutation: a descriptive study. Neuropsychology. 2004;18:658–664. - PubMed
-
- Berry G. Is prenatal myo-inositol deficiency a mechanism of CNS injury in galactosemia? J Inherit Metab Dis. 2011;34 - PubMed
-
- Berry GT, Nissim I, Lin Z, Mazur AT, Gibson JB, Segal S. Endogenous synthesis of galactose in normal men and patients with hereditary galactosemia. Lancet. 1995;346:1073–1074. - PubMed
-
- Berry GT, Reynolds RA, Yager CT, Segal S. Extended [13C] galactose oxidation studies in patients with galactosemia. Mol Genet Metab. 2004;82:130–136. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
