

doi: 10.1186/gb-2012-13-3-r22.
An integrative probabilistic model for identification of structural variation in sequencing data
Affiliations
- PMID: 22452995
- PMCID: PMC3439973
- DOI: 10.1186/gb-2012-13-3-r22
Item in Clipboard
An integrative probabilistic model for identification of structural variation in sequencing data
Genome Biol.
2012.
Abstract
Paired-end sequencing is a common approach for identifying structural variation (SV) in genomes. Discrepancies between the observed and expected alignments indicate potential SVs. Most SV detection algorithms use only one of the possible signals and ignore reads with multiple alignments. This results in reduced sensitivity to detect SVs, especially in repetitive regions. We introduce GASVPro, an algorithm combining both paired read and read depth signals into a probabilistic model which can analyze multiple alignments of reads. GASVPro outperforms existing methods with a 50-90% improvement in specificity on deletions and a 50% improvement on inversions.
Figures

Signals of structural variation from paired-end sequencing. (a) Fragments (black arches) from a test genome are sequenced from both ends and the resulting paired reads are mapped to a reference genome. Fragments containing the breakpoint of a structural variant (black arches with arrows) have a discordant mapping (red). (b) The GASV program [33] efficiently clusters discordant fragments supporting the same variant and provides precise information about the localization of the adjacency, (a,b), created by the rearrangement. For example, on the left a deletion of the interval from the reference creates a novel adjacency of breakends and . GASV represents the novel adjacency as a breakend polygon (shaded red trapezoid) where the left and right breakends of the variant must lie within the breakend polygon. In this example, we show breakend polygons for a deletion (left) and an inversion (right), each supported by two discordant fragments. (c) The presence of a structural variant is also indicated by changes in the depth of coverage of concordant mappings. For the deletion (left) the depth of coverage is low throughout the entire region, while for the inversion (right) the depth of coverage drops only near the breakends.
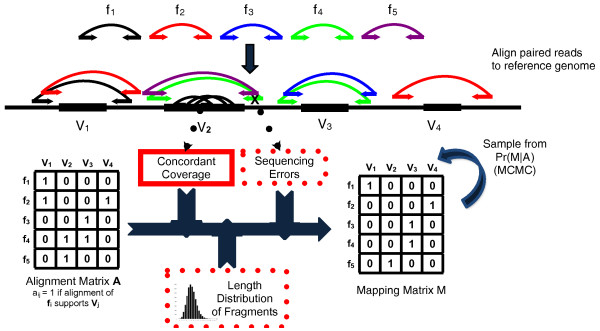
Overview of the GASVPro. Fragments  from a test genome are sequenced and the resulting paired reads are aligned to the reference. A fragment may either have a unique mapping or be ambiguous with multiple alignments to the reference. Following clustering of alignments (with GASV), the set
from a test genome are sequenced and the resulting paired reads are aligned to the reference. A fragment may either have a unique mapping or be ambiguous with multiple alignments to the reference. Following clustering of alignments (with GASV), the set  of possible structural variants and the fragments whose alignments support these variants are recorded in the alignment matrix A. As each fragment originates from a single location in the test genome, a fragment supports at most one structural variant. Thus, the mapping matrix M records the 'true' mapping for each fragment. GASVPro scores mapping matrices according to a generative probabilistic model that incorporates concordant mappings. GASVPro utilizes an MCMC procedure to efficiently sample over the space of possible mapping matrices defined by the alignment matrix A. The underlying probabilistic model can be easily generalized to consider additional features indicative of a 'true' mapping, such as the empirical fragment length distribution or probability of sequencing errors.
of possible structural variants and the fragments whose alignments support these variants are recorded in the alignment matrix A. As each fragment originates from a single location in the test genome, a fragment supports at most one structural variant. Thus, the mapping matrix M records the 'true' mapping for each fragment. GASVPro scores mapping matrices according to a generative probabilistic model that incorporates concordant mappings. GASVPro utilizes an MCMC procedure to efficiently sample over the space of possible mapping matrices defined by the alignment matrix A. The underlying probabilistic model can be easily generalized to consider additional features indicative of a 'true' mapping, such as the empirical fragment length distribution or probability of sequencing errors.
from a test genome are sequenced and the resulting paired reads are aligned to the reference. A fragment may either have a unique mapping or be ambiguous with multiple alignments to the reference. Following clustering of alignments (with GASV), the set of possible structural variants and the fragments whose alignments support these variants are recorded in the alignment matrix A. As each fragment originates from a single location in the test genome, a fragment supports at most one structural variant. Thus, the mapping matrix M records the 'true' mapping for each fragment. GASVPro scores mapping matrices according to a generative probabilistic model that incorporates concordant mappings. GASVPro utilizes an MCMC procedure to efficiently sample over the space of possible mapping matrices defined by the alignment matrix A. The underlying probabilistic model can be easily generalized to consider additional features indicative of a 'true' mapping, such as the empirical fragment length distribution or probability of sequencing errors.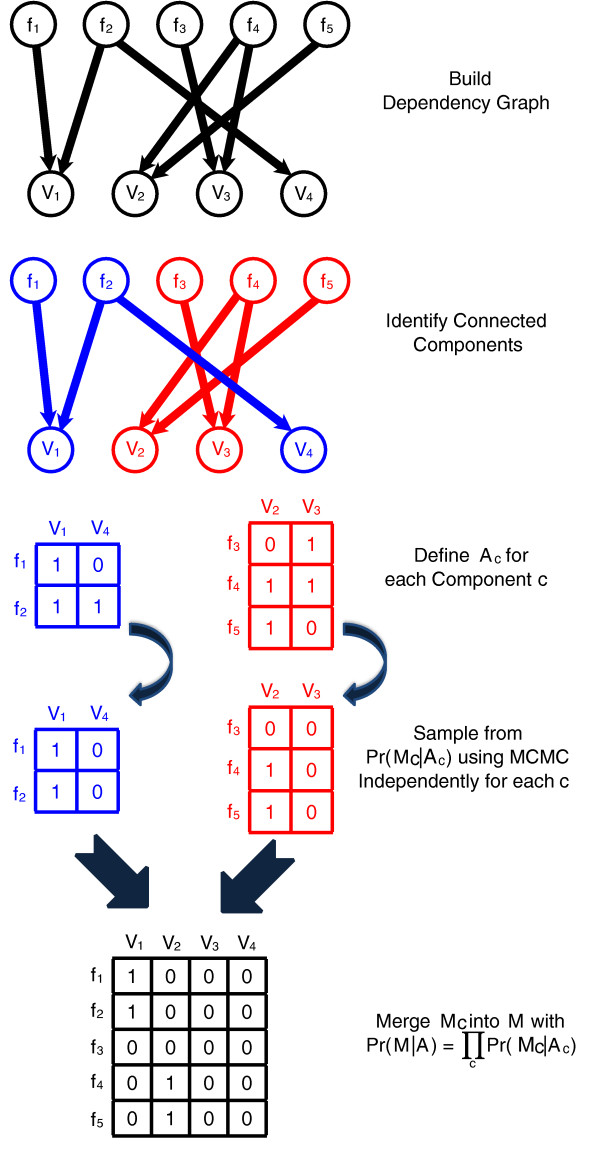
Sampling over connected components of alignments. The probabilistic model P(M|A) used by GASVPro allows for efficient decomposition of the original space of mapping matrices M into independent components. Thus, we sample using MCMC on each component independently and merge the results.
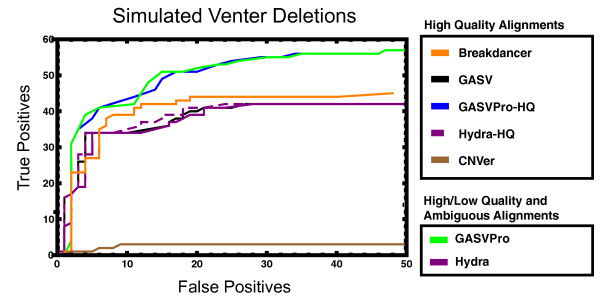
Simulated Venter chromosome 17. ROC curves comparing deletion predictions for Breakdancer, GASV, GASVPro-HQ, Hydra-HQ, CNVer, GASVPro and Hydra to the 124 deletions from Venter chromosome 17 with minimum deletion length 125 bp. All methods analyzed the same set of high-quality unique mappings; in addition, GASVPro and Hydra considered a set of lower-quality alignments, including ambiguous fragments with multiple alignments. Predictions of all methods were post-processed in an identical fashion and the resulting predictions were compared to the known coordinates for the Venter deletions according to the double uncertainty metric (see Methods).
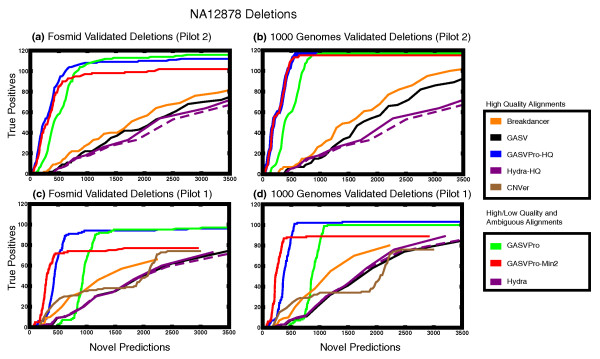
An 'ROC curve' comparing the number of known deletions that were correctly predicted (true positives) and the number of novel deletion predictions using sequencing data and validated deletions from the individual NA12878 in Pilot 1 and Pilot 2 of the 1000 Genomes Project. (As discussed in the Methods section, we separately considered two sets of validated deletions [16,44].)
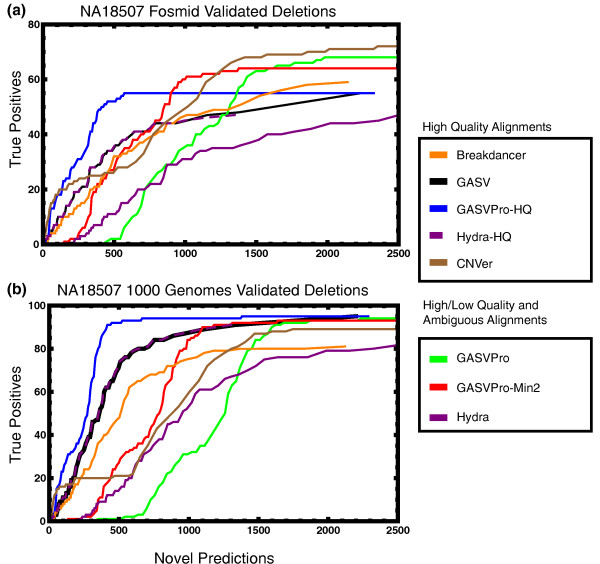
An 'ROC curve' comparing the number of known deletions and novel deletion predictions for NA18507. (As for individual NA12878, we separately considered two sets of validated deletions [16,44].)
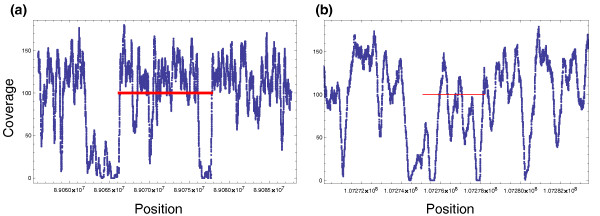
Concordant coverage per position for two known inversions successfully predicted by GASVPro. (a) A prediction with 99 discordant fragments overlaps a chromosome 4 inversion with left breakend uncertainty of 89.040 to 89.069 Mb and right breakend uncertainty of 89.075 to 89.108 Mb. (b) A prediction with 15 discordant fragments overlaps a chromosome 6 inversion with left breakend uncertainty of 107.245 to 107.283 Mb and right breakend uncertainty of 107.277 to 107.315 Mb. For both predictions, a thick red line indicates the minimum and maximum mapped ends (x,y) for all supporting discordant fragments.
References
-
- Conrad DF, Pinto D, Redon R, Feuk L, Gokcumen O, Zhang Y, Aerts J, Andrews TD, Barnes C, Campbell P, Fitzgerald T, Hu M, Ihm CH, Kristiansson K, Macarthur DG, Macdonald JR, Onyiah I, Pang AW, Robson S, Stirrups K, Valsesia A, Walter K, Wei J. Wellcome Trust Case Control Consortium. Tyler-Smith C, Carter NP, Lee C, Scherer SW, Hurles ME. Origins and functional impact of copy number variation in the human genome. Nature. 2009;464:704–712. - PMC - PubMed
-
- Ding L, Ellis MJ, Li S, Larson DE, Chen K, Wallis JW, Harris CC, McLellan MD, Fulton RS, Fulton LL, Abbott RM, Hoog J, Dooling DJ, Koboldt DC, Schmidt H, Kalicki J, Zhang Q, Chen L, Lin L, Wendl MC, McMichael JF, Magrini VJ, Cook L, McGrath SD, Vickery TL, Appelbaum E, Deschryver K, Davies S, Guintoli T, Lin L. et al.Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. - DOI - PMC - PubMed
-
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordóñez GR, Bignell GR, Ye K, Alipaz J, Bauer MJ, Beare D, Butler A, Carter RJ, Chen L, Cox AJ, Edkins S, Kokko-Gonzales PI, Gormley NA, Grocock RJ, Haudenschild CD, Hims MM, James T, Jia M, Kingsbury Z, Leroy C, Marshall J, Menzies A. et al.A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2009;463:191–196. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
