

Misfolded PrP and a novel mechanism of proteasome inhibition
- PMID: 22453175
- PMCID: PMC3338962
- DOI: 10.4161/pri.6.1.18272
Misfolded PrP and a novel mechanism of proteasome inhibition
Abstract
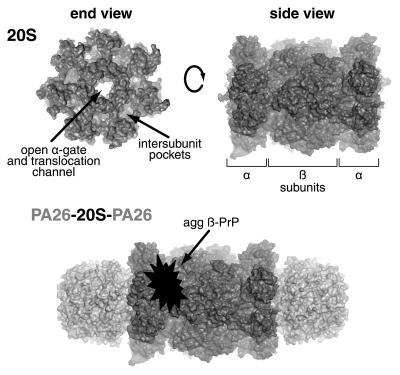
Prion diseases comprise a family of fatal neurodegenerative disorders caused by the conformational re-arrangement of a normal host-encoded protein, PrP (C) , to an abnormal infectious isoform termed PrP (Sc) . Currently, the precise cellular mechanism(s) underlying prion disease pathogenesis remain unclear. Evidence suggests a role for the ubiquitin proteasome system (UPS), a protein degradation pathway that is critical for maintaining cellular proteostasis. Dysfunction of the UPS has been implicated in various neurodegenerative diseases. However, the mechanisms of this impairment remain unknown in many cases, and evidence that disease-associated misfolded proteins are able to directly inhibit the function of the proteasome has been lacking. Recently, we have shown data describing a mechanism of proteasome impairment by the direct interaction of β-sheet-rich PrP to reduce gate opening and inhibit substrate entry. This novel mechanism may provide a model for how other misfolded, disease-associated proteins might interact with the proteasome to disrupt its function. Targeting the UPS to restore proteostasis in neurodegenerative disorders in which misfolded proteins accumulate offers a possible target for therapeutic intervention.
Figures
References
-
- Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials