

FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure
- PMID: 22453831
- PMCID: PMC3316891
- DOI: 10.1038/ncomms1755
FAD-dependent lysine-specific demethylase-1 regulates cellular energy expenditure
Abstract
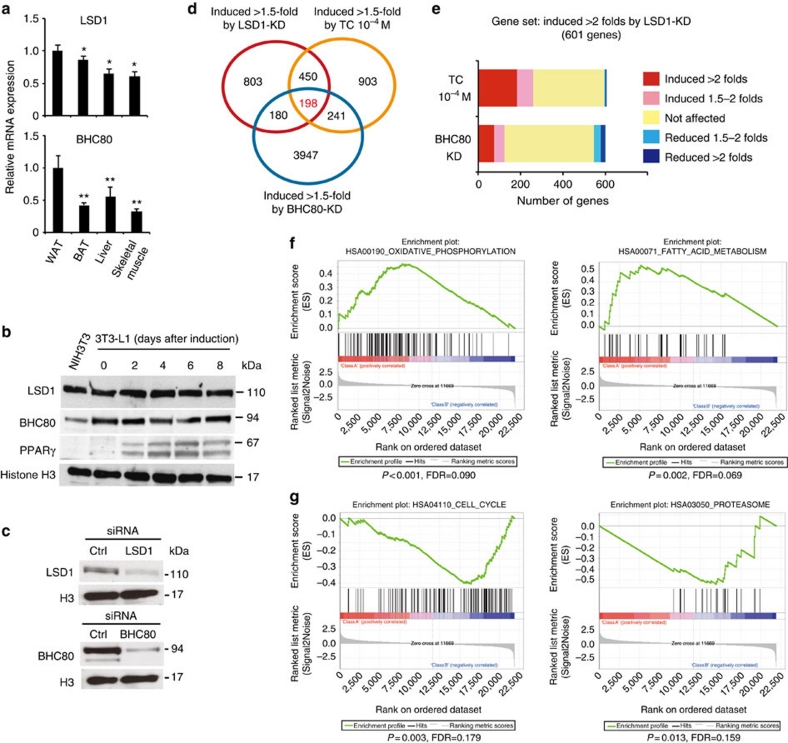
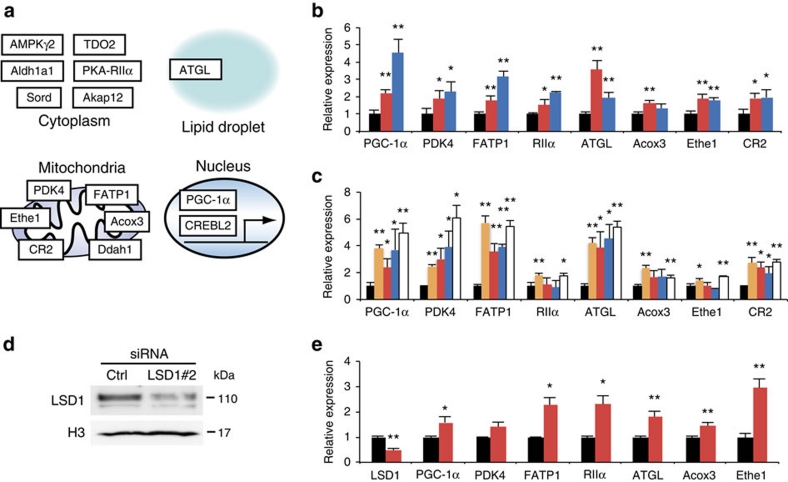
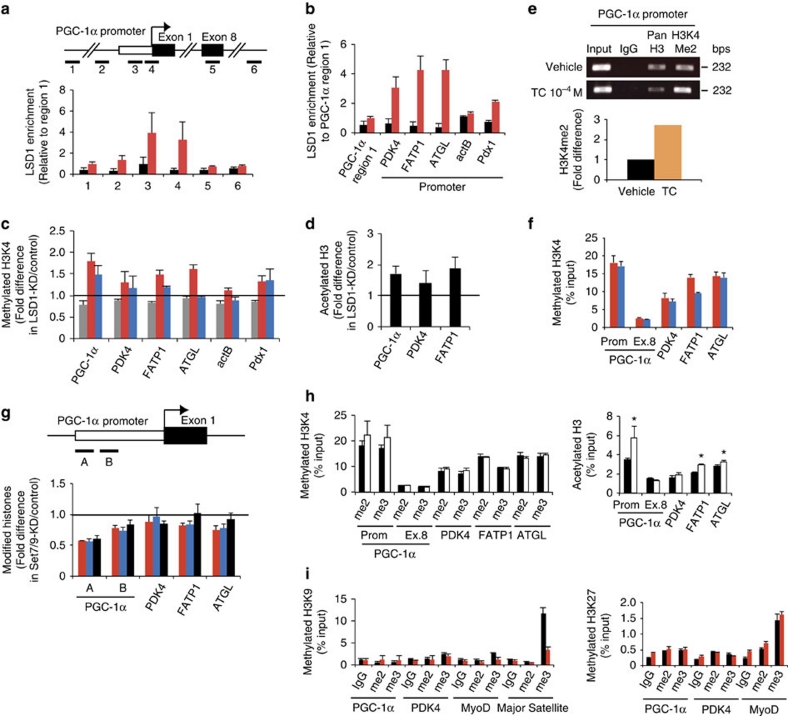
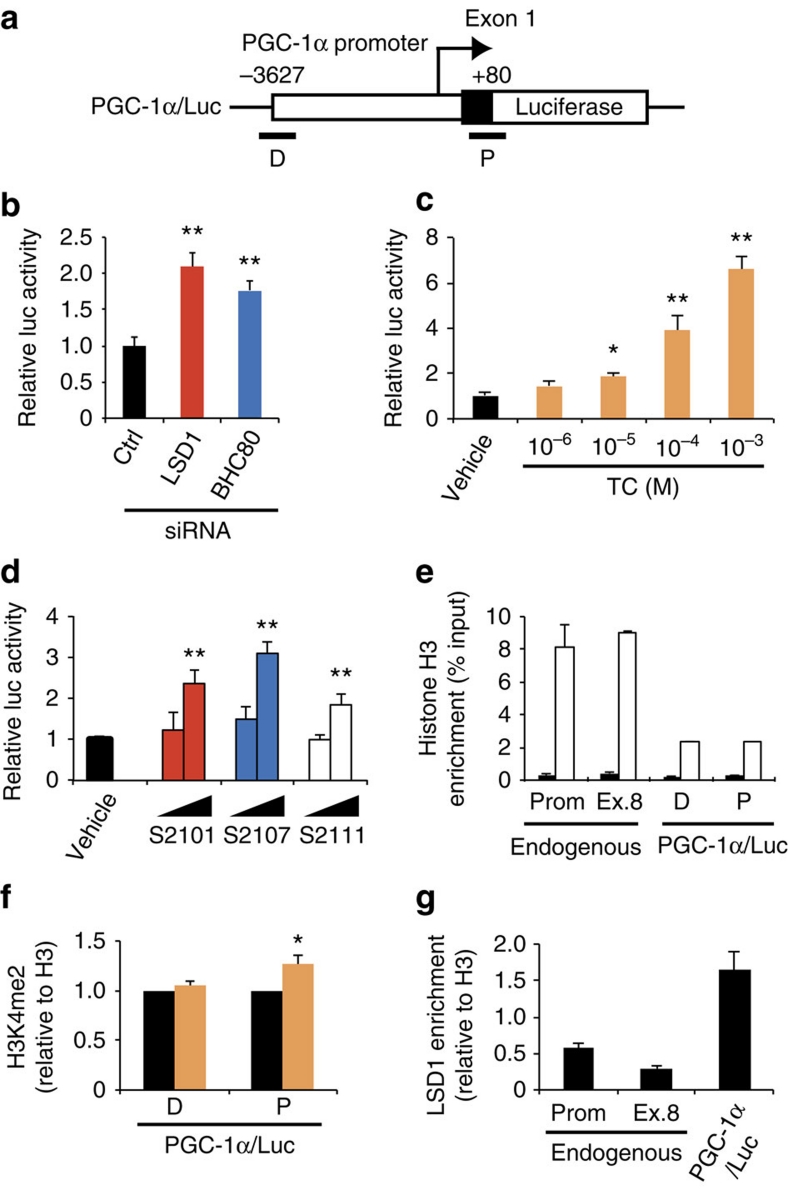
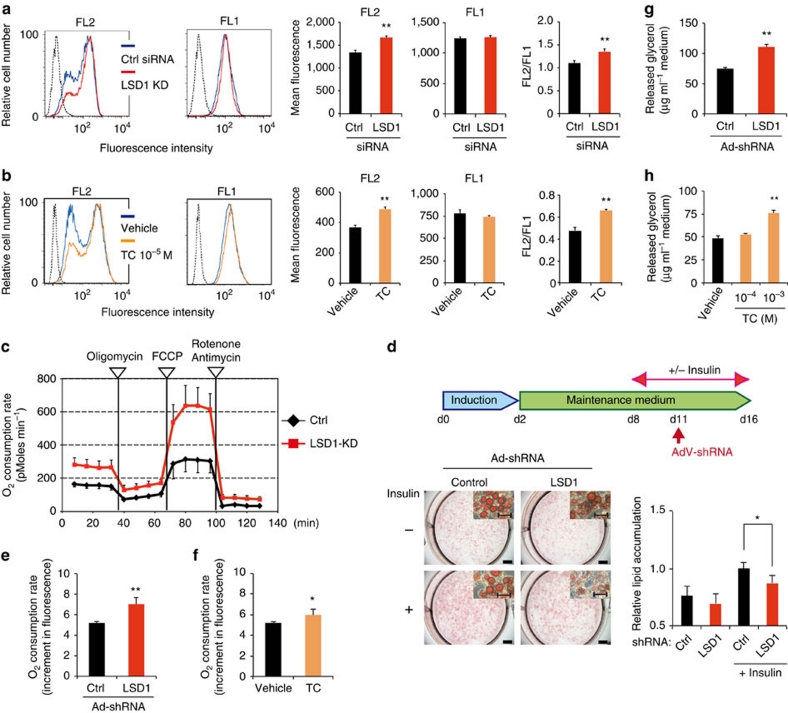
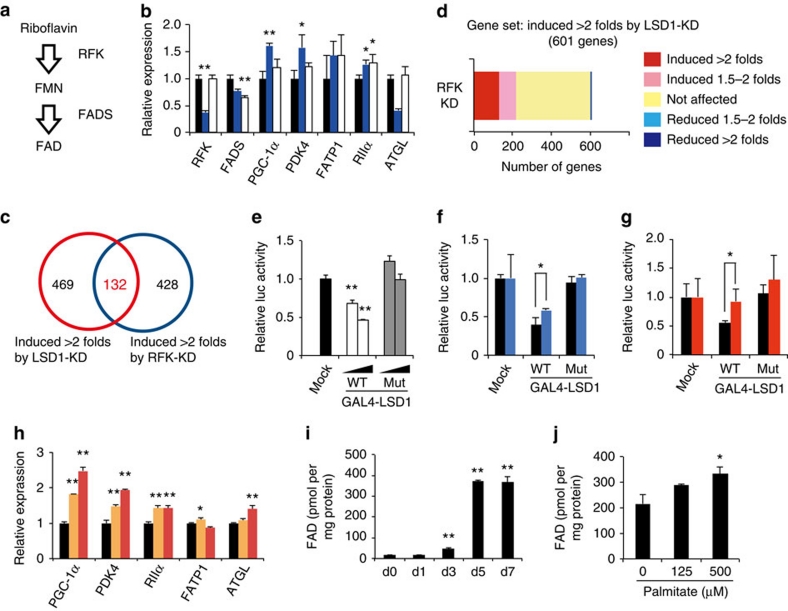
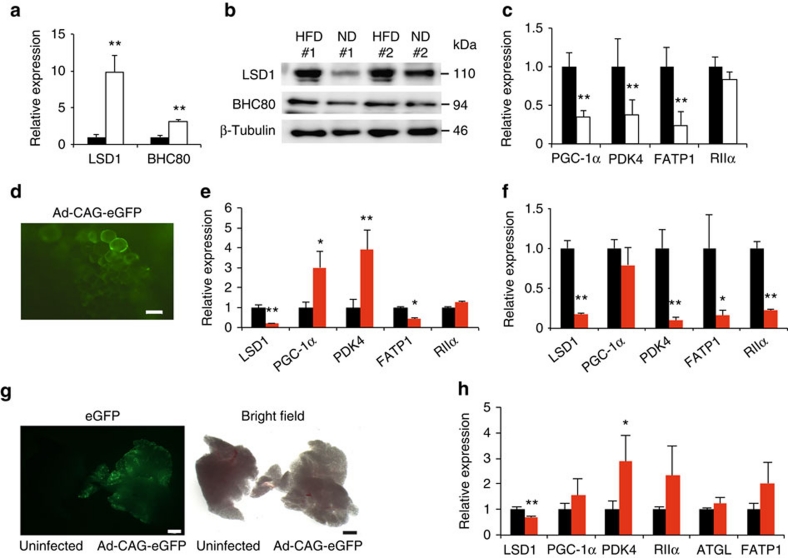
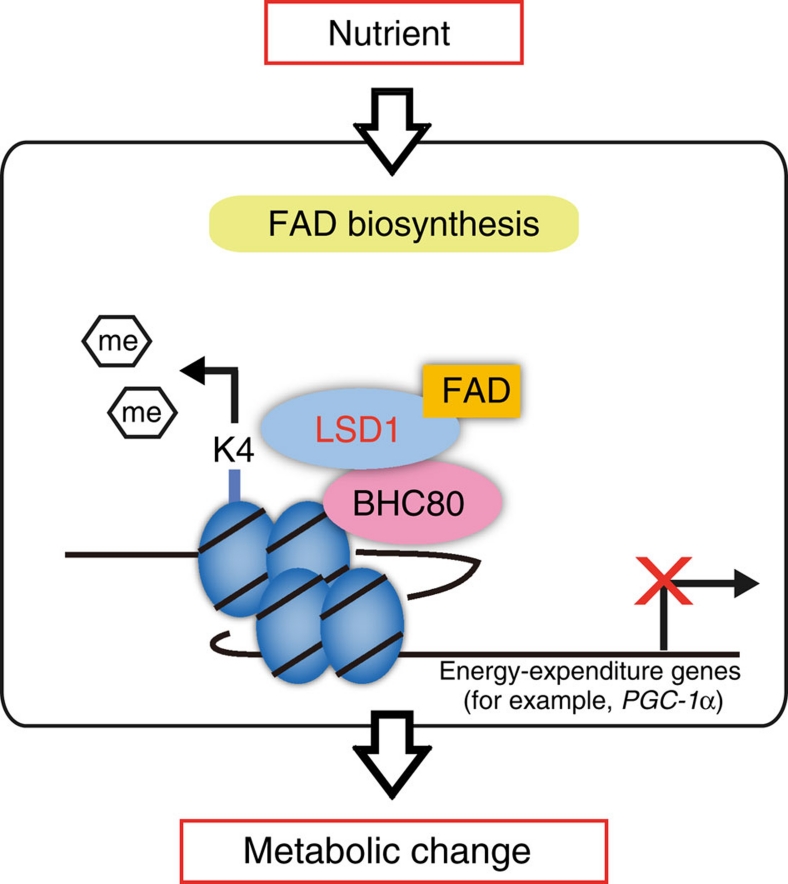
Environmental factors such as nutritional state may act on the epigenome that consequently contributes to the metabolic adaptation of cells and the organisms. The lysine-specific demethylase-1 (LSD1) is a unique nuclear protein that utilizes flavin adenosine dinucleotide (FAD) as a cofactor. Here we show that LSD1 epigenetically regulates energy-expenditure genes in adipocytes depending on the cellular FAD availability. We find that the loss of LSD1 function, either by short interfering RNA or by selective inhibitors in adipocytes, induces a number of regulators of energy expenditure and mitochondrial metabolism such as PPARγ coactivator-1α resulting in the activation of mitochondrial respiration. In the adipose tissues from mice on a high-fat diet, expression of LSD1-target genes is reduced, compared with that in tissues from mice on a normal diet, which can be reverted by suppressing LSD1 function. Our data suggest a novel mechanism where LSD1 regulates cellular energy balance through coupling with cellular FAD biosynthesis.
Figures
References
-
- Felsenfeld G. & Groudine M. Controlling the double helix. Nature 421, 448–453 (2003). - PubMed
-
- Gluckman P. D. & Hanson M. A. The developmental origins of the metabolic syndrome. Trends Endocrinol. Metab. 15, 183–187 (2004). - PubMed
-
- Gallou-Kabani C. & Junien C. Nutritional epigenomics of metabolic syndrome: new perspective against the epidemic. Diabetes 54, 1899–1906 (2005). - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
