

A quantitative atlas of polyadenylation in five mammals
- PMID: 22454233
- PMCID: PMC3371698
- DOI: 10.1101/gr.132563.111
A quantitative atlas of polyadenylation in five mammals
Abstract
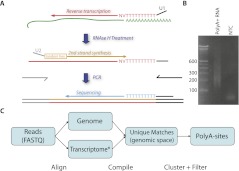
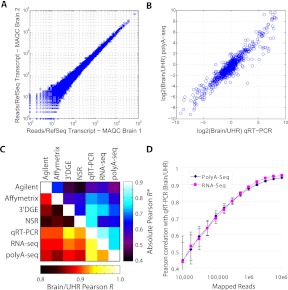
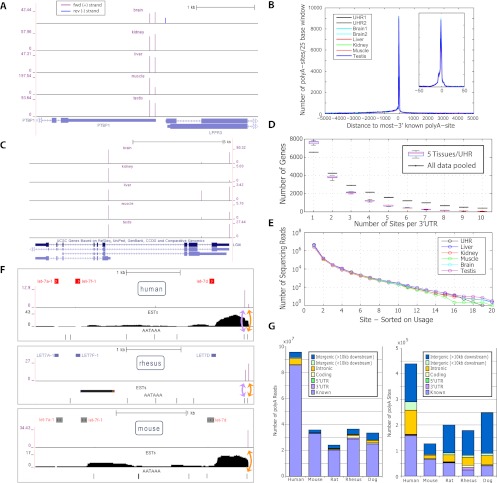
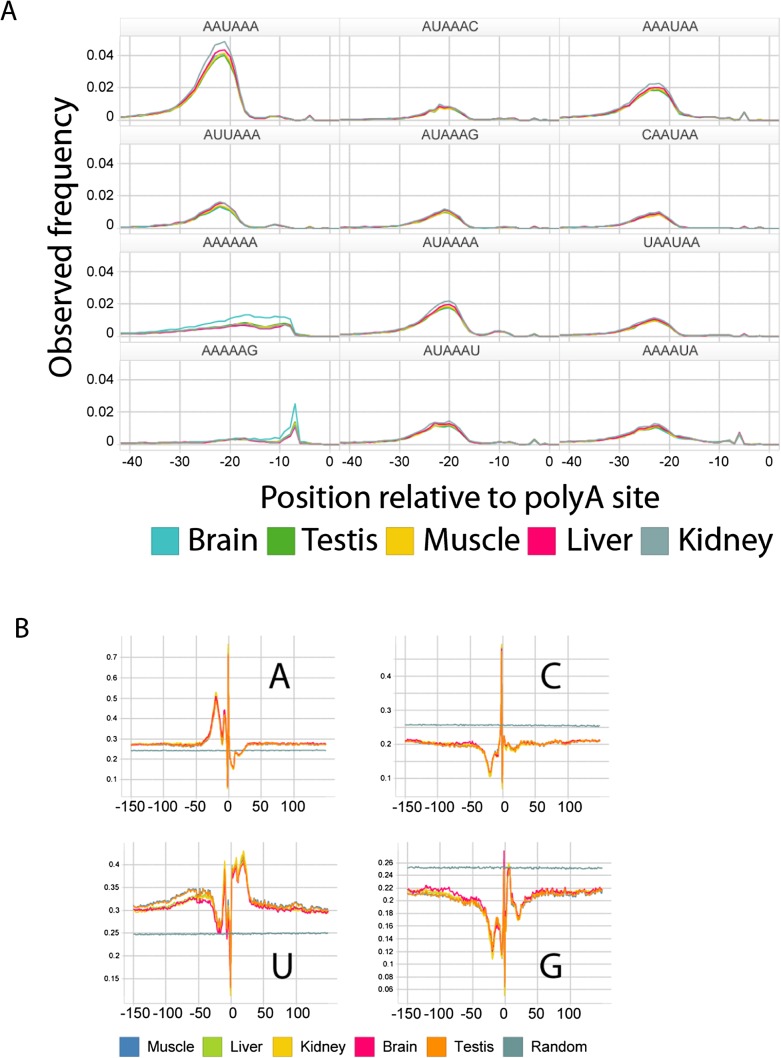
We developed PolyA-seq, a strand-specific and quantitative method for high-throughput sequencing of 3' ends of polyadenylated transcripts, and used it to globally map polyadenylation (polyA) sites in 24 matched tissues in human, rhesus, dog, mouse, and rat. We show that PolyA-seq is as accurate as existing RNA sequencing (RNA-seq) approaches for digital gene expression (DGE), enabling simultaneous mapping of polyA sites and quantitative measurement of their usage. In human, we confirmed 158,533 known sites and discovered 280,857 novel sites (FDR < 2.5%). On average 10% of novel human sites were also detected in matched tissues in other species. Most novel sites represent uncharacterized alternative polyA events and extensions of known transcripts in human and mouse, but primarily delineate novel transcripts in the other three species. A total of 69.1% of known human genes that we detected have multiple polyA sites in their 3'UTRs, with 49.3% having three or more. We also detected polyadenylation of noncoding and antisense transcripts, including constitutive and tissue-specific primary microRNAs. The canonical polyA signal was strongly enriched and positionally conserved in all species. In general, usage of polyA sites is more similar within the same tissues across different species than within a species. These quantitative maps of polyA usage in evolutionarily and functionally related samples constitute a resource for understanding the regulatory mechanisms underlying alternative polyadenylation.
Figures
References
-
- Armour CD, Castle JC, Chen R, Babak T, Loerch P, Jackson S, Shah JK, Dey J, Rohl CA, Johnson JM, et al. 2009. Digital transcriptome profiling using selective hexamer priming for cDNA synthesis. Nat Methods 6: 647–649 - PubMed
-
- Asmann YW, Klee EW, Thompson EA, Perez EA, Middha S, Oberg AL, Therneau TM, Smith DI, Poland GA, Wieben ED, et al. 2009. 3′ tag digital gene expression profiling of human brain and universal reference RNA using Illumina Genome Analyzer. BMC Genomics 10: 531 doi: 10.1186/1471-2164-10-531 - PMC - PubMed
-
- Boguski MS, Lowe TM, Tolstoshev CM 1993. dbEST–database for “expressed sequence tags.” Nat Genet 4: 332–333 - PubMed
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases