

Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation
- PMID: 22454539
- PMCID: PMC3418851
- DOI: 10.1074/mcp.M112.017707
Platform-independent and label-free quantitation of proteomic data using MS1 extracted ion chromatograms in skyline: application to protein acetylation and phosphorylation
Abstract
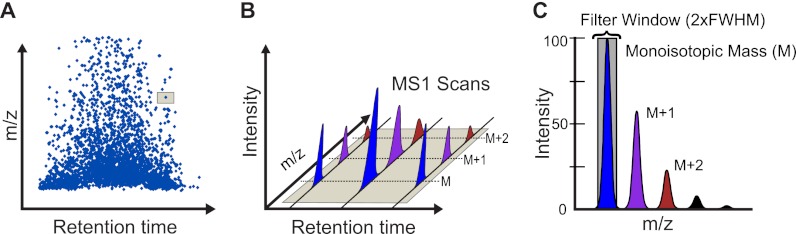
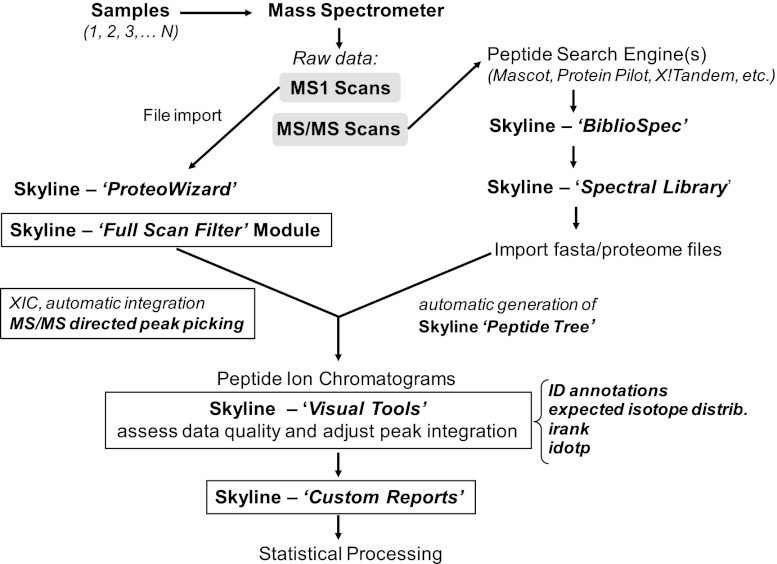
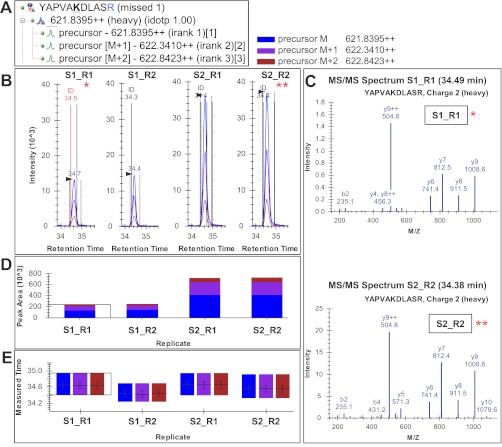
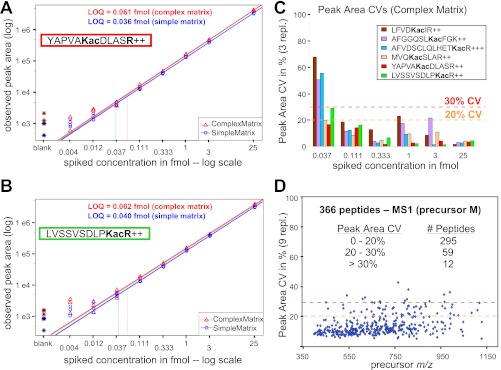
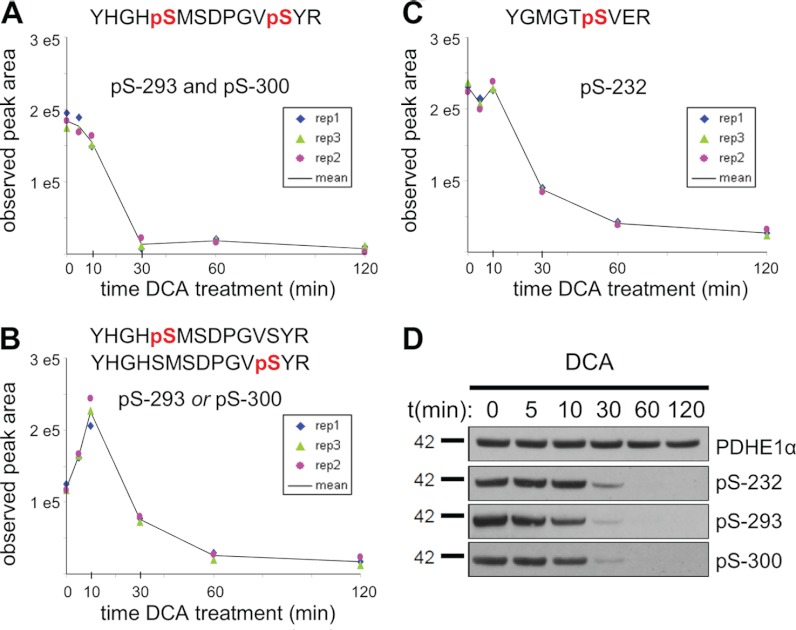
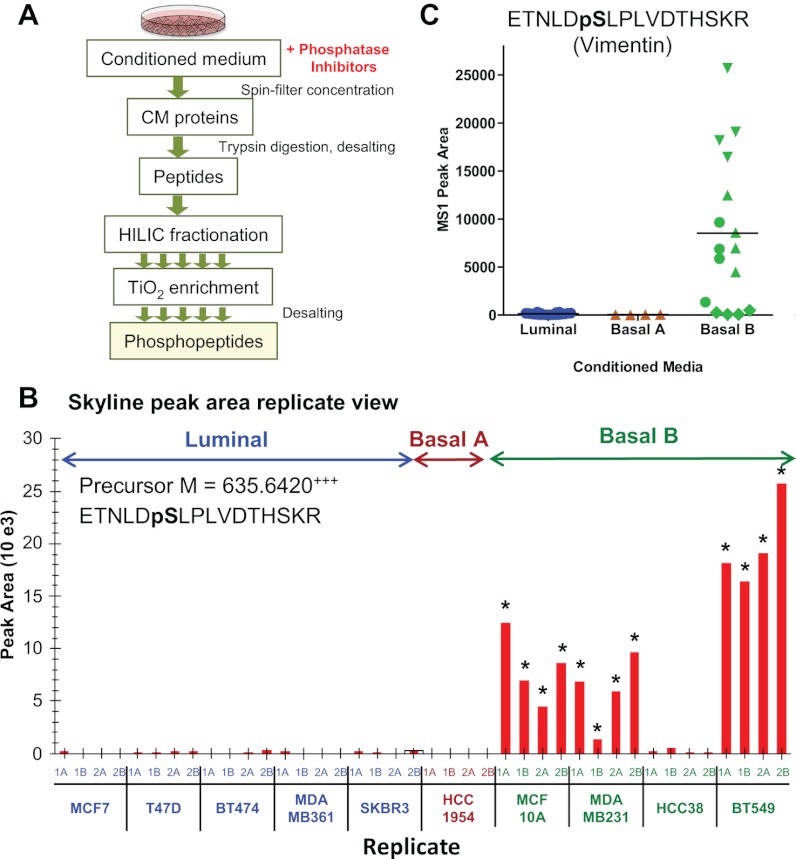
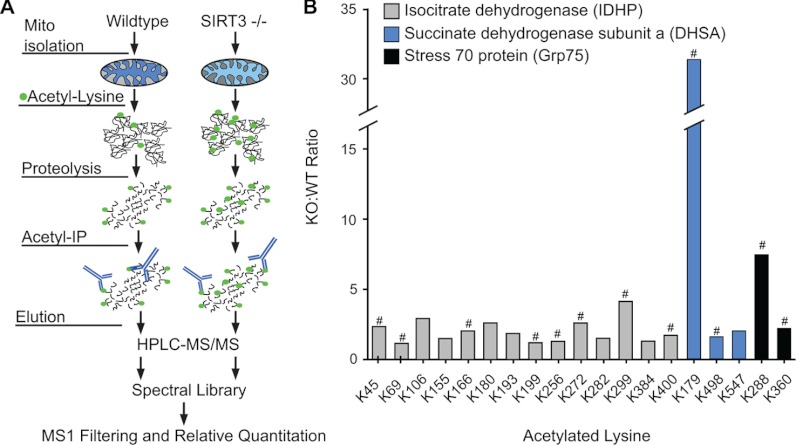
Despite advances in metabolic and postmetabolic labeling methods for quantitative proteomics, there remains a need for improved label-free approaches. This need is particularly pressing for workflows that incorporate affinity enrichment at the peptide level, where isobaric chemical labels such as isobaric tags for relative and absolute quantitation and tandem mass tags may prove problematic or where stable isotope labeling with amino acids in cell culture labeling cannot be readily applied. Skyline is a freely available, open source software tool for quantitative data processing and proteomic analysis. We expanded the capabilities of Skyline to process ion intensity chromatograms of peptide analytes from full scan mass spectral data (MS1) acquired during HPLC MS/MS proteomic experiments. Moreover, unlike existing programs, Skyline MS1 filtering can be used with mass spectrometers from four major vendors, which allows results to be compared directly across laboratories. The new quantitative and graphical tools now available in Skyline specifically support interrogation of multiple acquisitions for MS1 filtering, including visual inspection of peak picking and both automated and manual integration, key features often lacking in existing software. In addition, Skyline MS1 filtering displays retention time indicators from underlying MS/MS data contained within the spectral library to ensure proper peak selection. The modular structure of Skyline also provides well defined, customizable data reports and thus allows users to directly connect to existing statistical programs for post hoc data analysis. To demonstrate the utility of the MS1 filtering approach, we have carried out experiments on several MS platforms and have specifically examined the performance of this method to quantify two important post-translational modifications: acetylation and phosphorylation, in peptide-centric affinity workflows of increasing complexity using mouse and human models.
Figures
References
-
- Bantscheff M., Schirle M., Sweetman G., Rick J., Kuster B. (2007) Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanal. Chem. 389, 1017–1031 - PubMed
-
- Mueller L. N., Brusniak M. Y., Mani D. R., Aebersold R. (2008) An assessment of software solutions for the analysis of mass spectrometry based quantitative proteomics data. J. Proteome Res. 7, 51–61 - PubMed
-
- Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 - PubMed
-
- Krüger M., Moser M., Ussar S., Thievessen I., Luber C. A., Forner F., Schmidt S., Zanivan S., Fässler R., Mann M. (2008) SILAC mouse for quantitative proteomics uncovers kindlin-3 as an essential factor for red blood cell function. Cell 134, 353–364 - PubMed
-
- Zanivan S., Krueger M., Mann M. (2012) In vivo quantitative proteomics: The SILAC mouse. Methods Mol. Biol. 757, 435–450 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
