

Identifying individual DNA species in a complex mixture by precisely measuring the spacing between nicking restriction enzymes with atomic force microscope
- PMID: 22456455
- PMCID: PMC3405743
- DOI: 10.1098/rsif.2012.0024
Identifying individual DNA species in a complex mixture by precisely measuring the spacing between nicking restriction enzymes with atomic force microscope
Abstract
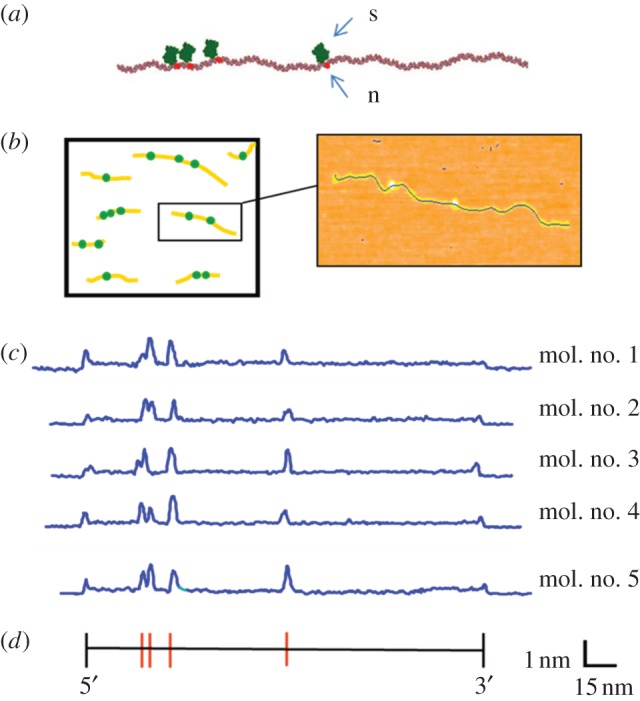
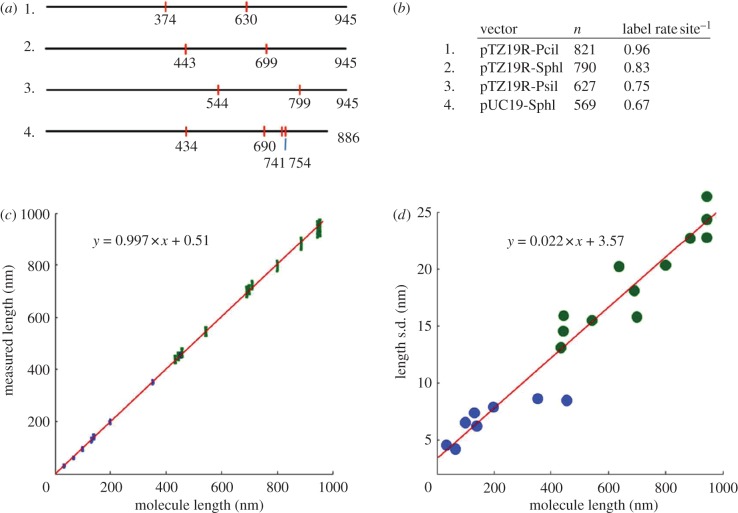
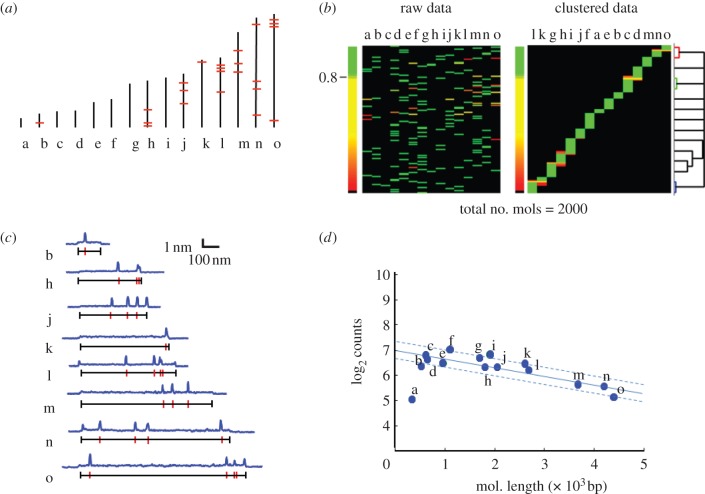
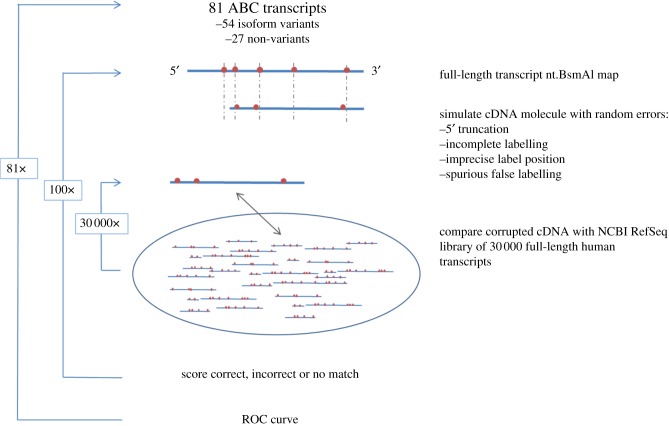
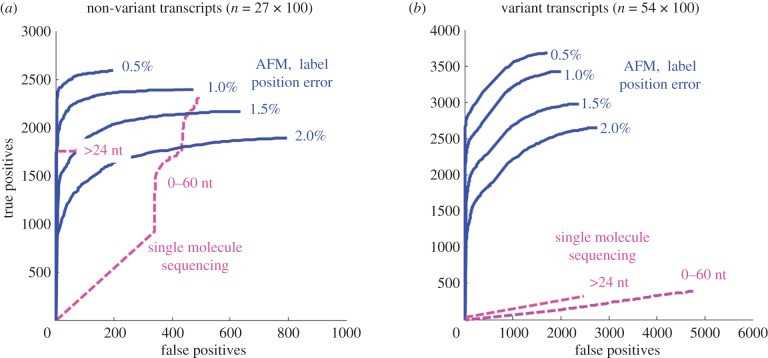
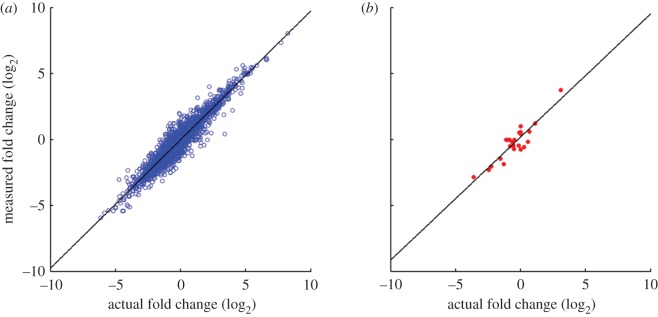
We discuss a novel atomic force microscope-based method for identifying individual short DNA molecules (<5000 bp) within a complex mixture by measuring the intra-molecular spacing of a few sequence-specific topographical labels in each molecule. Using this method, we accurately determined the relative abundance of individual DNA species in a 15-species mixture, with fewer than 100 copies per species sampled. To assess the scalability of our approach, we conducted a computer simulation, with realistic parameters, of the hypothetical problem of detecting abundance changes in individual gene transcripts between two single-cell human messenger RNA samples, each containing roughly 9000 species. We found that this approach can distinguish transcript species abundance changes accurately in most cases, including transcript isoforms which would be challenging to quantitate with traditional methods. Given its sensitivity and procedural simplicity, our approach could be used to identify transcript-derived complementary DNAs, where it would have substantial technical and practical advantages versus established techniques in situations where sample material is scarce.
Figures
References
-
- Burbulis I., Yamaguchi K., Gordon A., Carlson R., Brent R. 2005. Using protein–DNA chimeras to detect and count small numbers of molecules. Nat. Methods 2, 31–3710.1038/nmeth729 (doi:10.1038/nmeth729) - DOI - DOI - PubMed
-
- Nygaard V., Hovig E. 2009. Methods for quantitation of gene expression. Front. Biosci. 14, 552–56910.2741/3262 (doi:10.2741/3262) - DOI - DOI - PubMed
-
- Nygaard V., Holden M., Loland A., Langaas M., Myklebost O., Hovig E. 2005. Limitations of mRNA amplification from small-size cell samples. BMC Genom. 6, 147.10.1186/1471-2164-6-147 (doi:10.1186/1471-2164-6-147) - DOI - DOI - PMC - PubMed
-
- Shi L. M., et al. 2006. The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 24, 1151–116110.1038/nbt1239 (doi:10.1038/nbt1239) - DOI - DOI - PMC - PubMed
-
- Bishop J. O., Morton J. G., Rosbash M., Richards M. 1974. Three abundance classes in HeLa-cell messenger-RNA. Nature 250, 199–20410.1038/250199a0 (doi:10.1038/250199a0) - DOI - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources