

Factors that control catalytic two- versus four-electron reduction of dioxygen by copper complexes
- PMID: 22462521
- PMCID: PMC3381452
- DOI: 10.1021/ja211656g
Factors that control catalytic two- versus four-electron reduction of dioxygen by copper complexes
Abstract
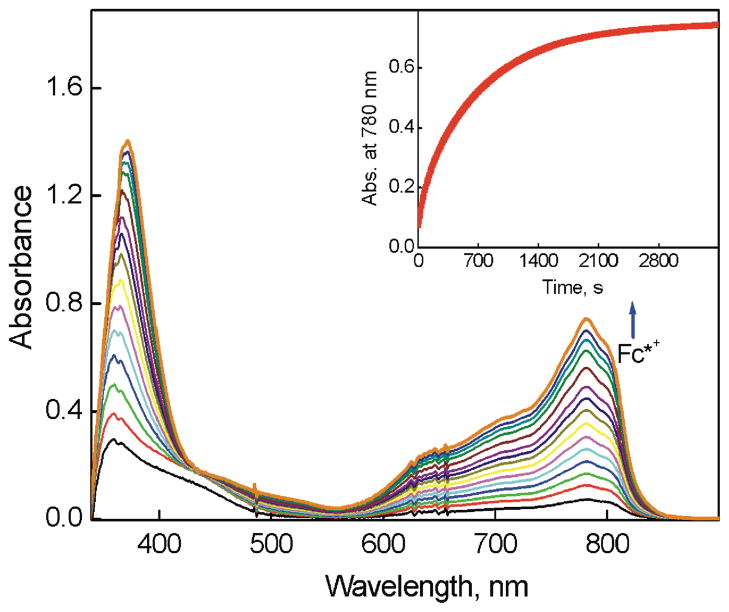
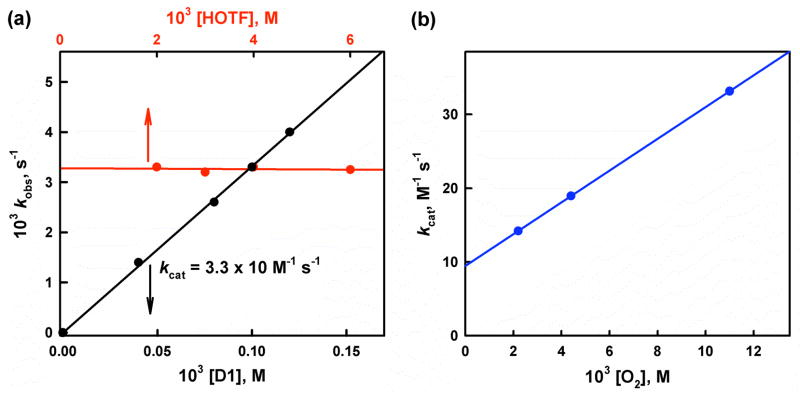
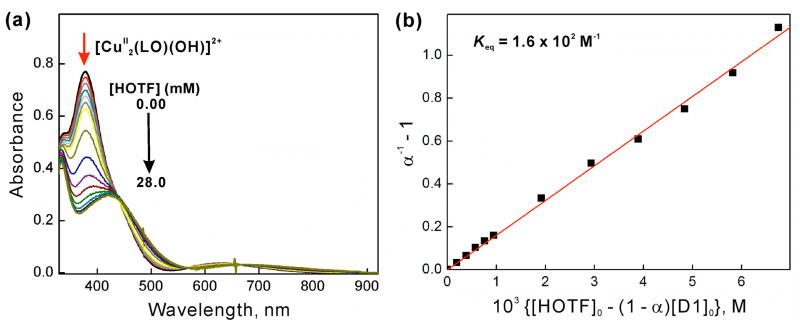
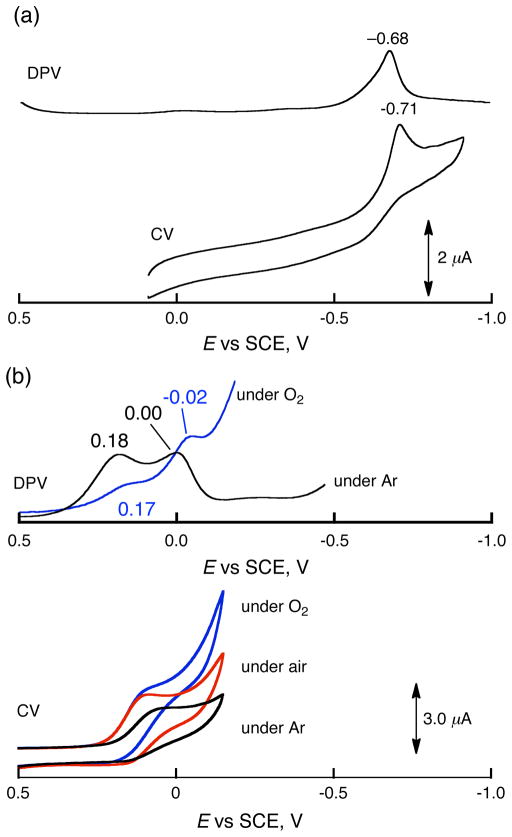
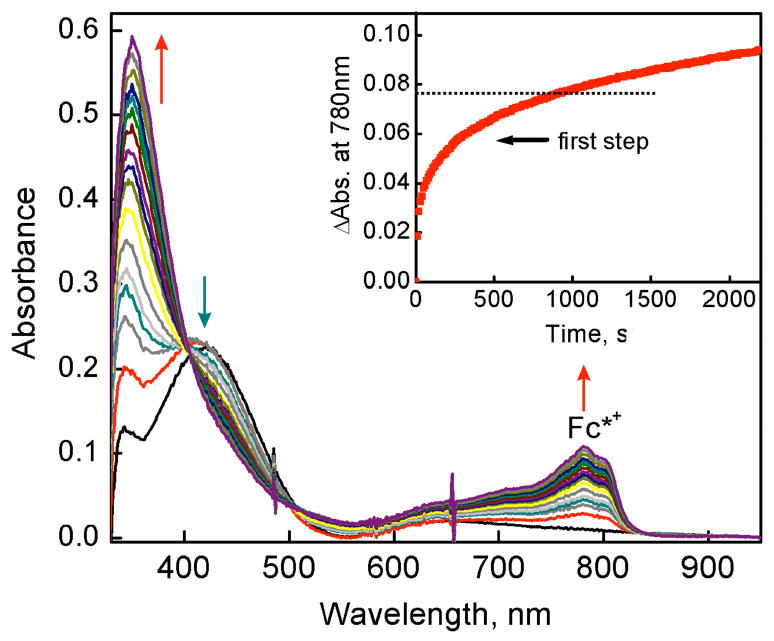
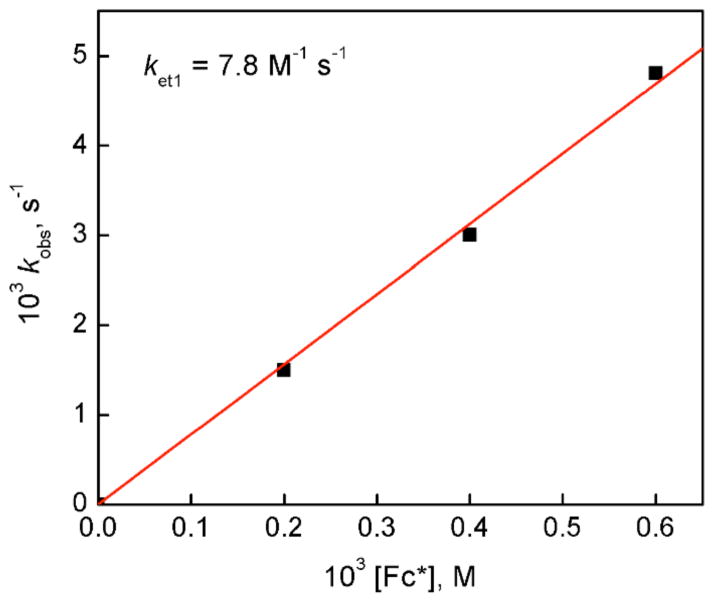
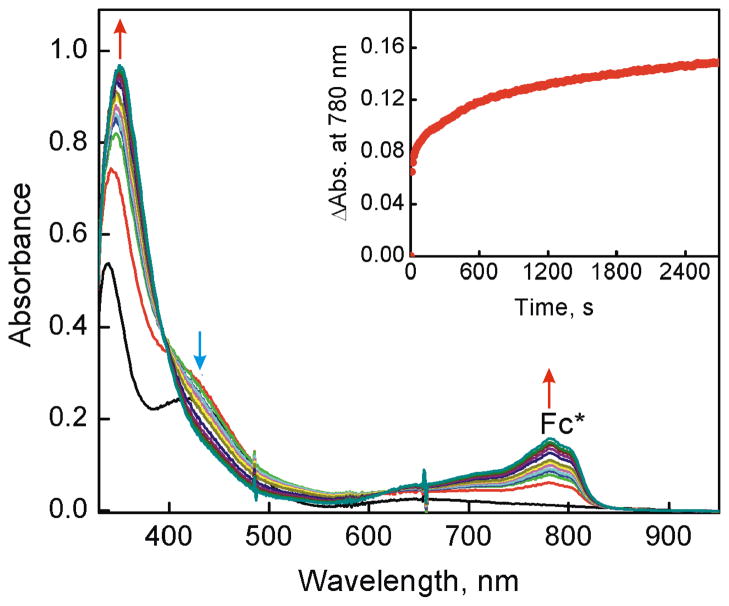
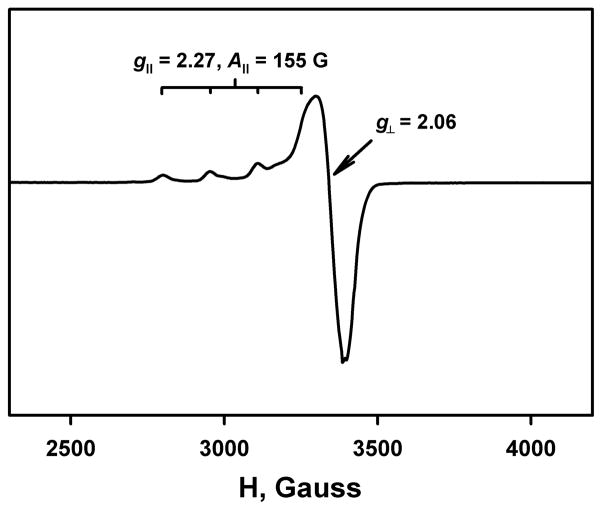
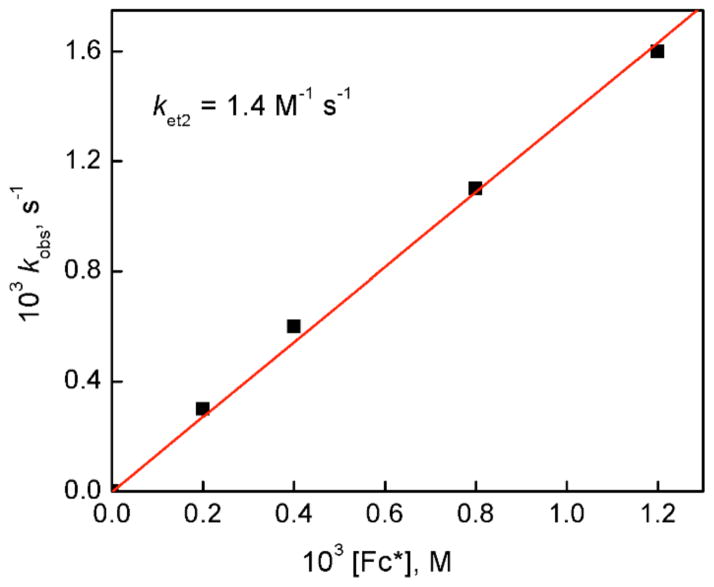
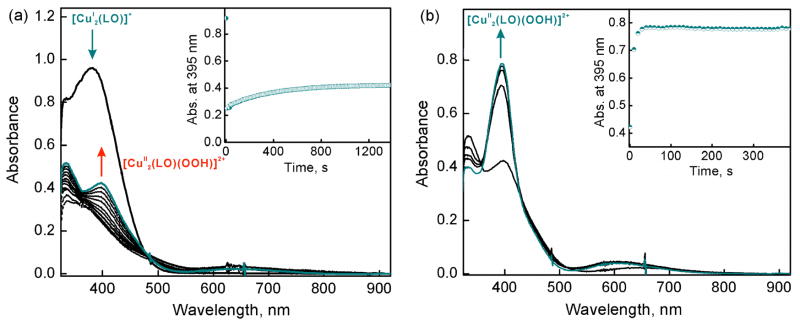
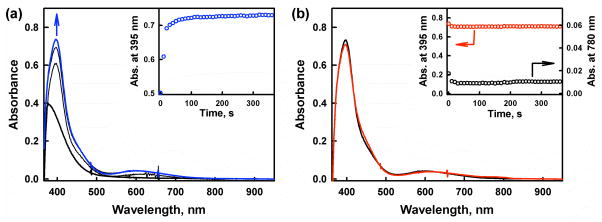
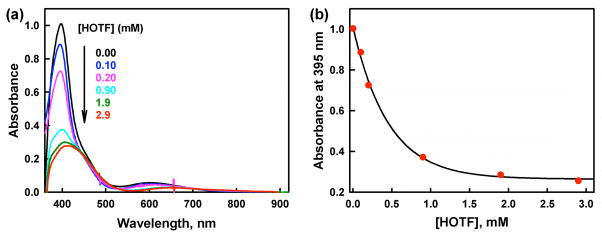
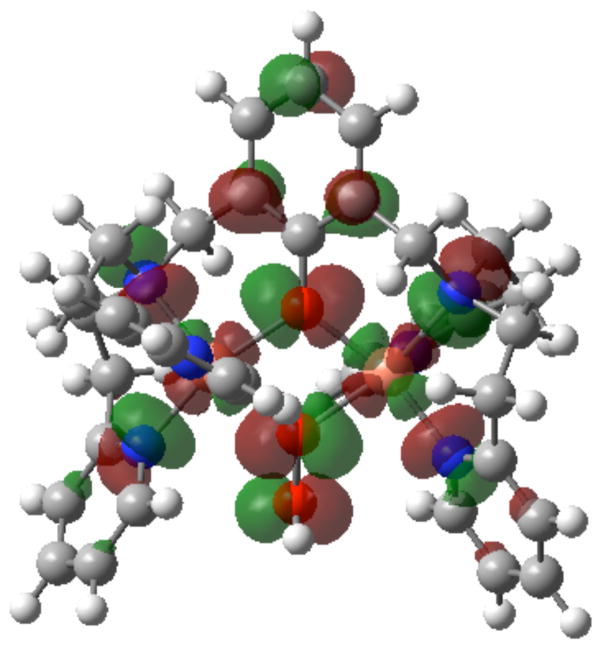
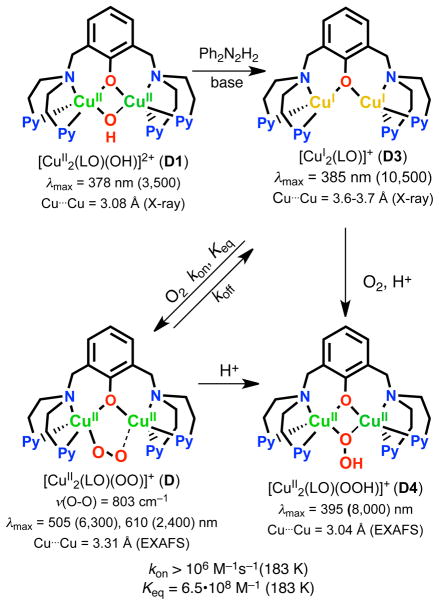
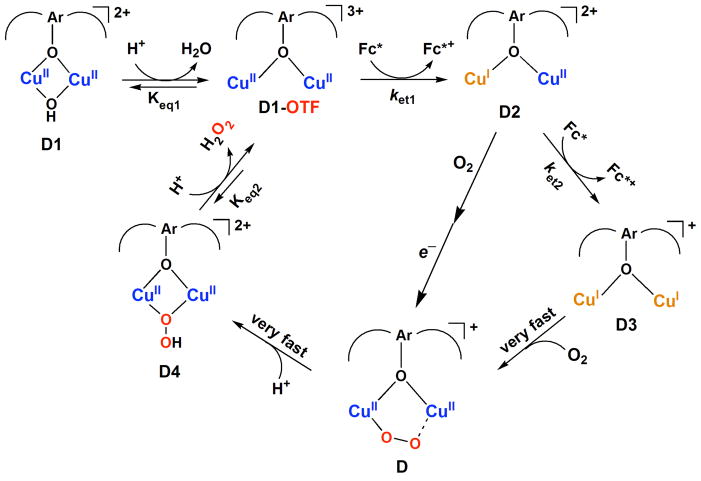
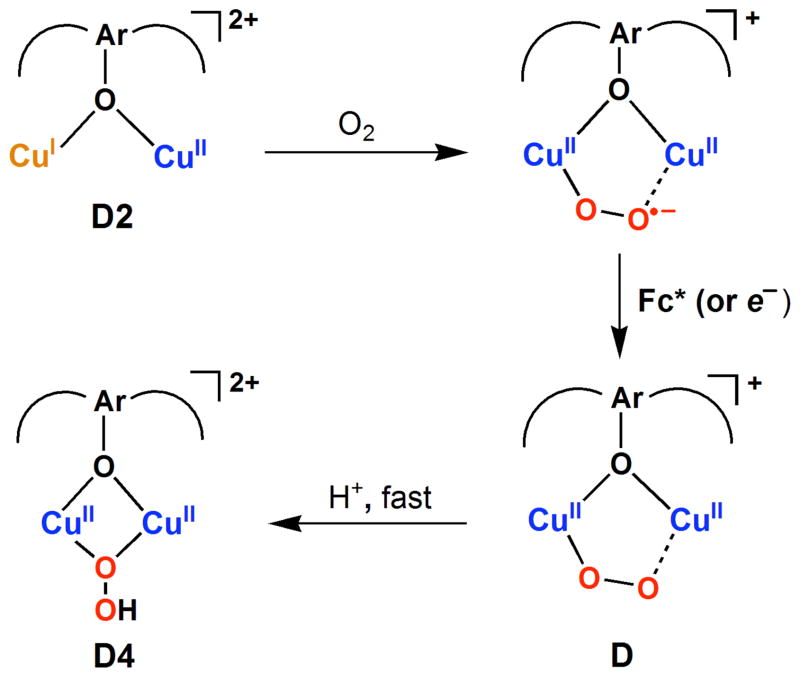
The selective two-electron reduction of O(2) by one-electron reductants such as decamethylferrocene (Fc*) and octamethylferrocene (Me(8)Fc) is efficiently catalyzed by a binuclear Cu(II) complex [Cu(II)(2)(LO)(OH)](2+) (D1) {LO is a binucleating ligand with copper-bridging phenolate moiety} in the presence of trifluoroacetic acid (HOTF) in acetone. The protonation of the hydroxide group of [Cu(II)(2)(LO)(OH)](2+) with HOTF to produce [Cu(II)(2)(LO)(OTF)](2+) (D1-OTF) makes it possible for this to be reduced by 2 equiv of Fc* via a two-step electron-transfer sequence. Reactions of the fully reduced complex [Cu(I)(2)(LO)](+) (D3) with O(2) in the presence of HOTF led to the low-temperature detection of the absorption spectra due to the peroxo complex [Cu(II)(2)(LO)(OO)] (D) and the protonated hydroperoxo complex [Cu(II)(2)(LO)(OOH)](2+) (D4). No further Fc* reduction of D4 occurs, and it is instead further protonated by HOTF to yield H(2)O(2) accompanied by regeneration of [Cu(II)(2)(LO)(OTF)](2+) (D1-OTF), thus completing the catalytic cycle for the two-electron reduction of O(2) by Fc*. Kinetic studies on the formation of Fc*(+) under catalytic conditions as well as for separate examination of the electron transfer from Fc* to D1-OTF reveal there are two important reaction pathways operating. One is a rate-determining second reduction of D1-OTF, thus electron transfer from Fc* to a mixed-valent intermediate [Cu(II)Cu(I)(LO)](2+) (D2), which leads to [Cu(I)(2)(LO)](+) that is coupled with O(2) binding to produce [Cu(II)(2)(LO)(OO)](+) (D). The other involves direct reaction of O(2) with the mixed-valent compound D2 followed by rapid Fc* reduction of a putative superoxo-dicopper(II) species thus formed, producing D.
Figures



References
-
- Solomon EI, Ginsbach JW, Heppner DE, Kieber-Emmons MT, Kjaergaard CH, Smeets PJ, Tian L, Woertink JS. Faraday Discuss. 2011;148:11–39. - PMC - PubMed
- Solomon EI, Sundaram UM, Machonkin TE. Chem Rev. 1996;96:2563–2605. - PubMed
- Solomon EI, Chen P, Metz M, Lee SK, Palmer AE. Angew Chem Int Ed. 2001;40:4570–4590. - PubMed
- Karlin KD, Tyeklár Z, editors. Bioinorganic Chemistry of Copper. Chapman & Hall; New York: 1993.
- Karlin KD, Zuberbühler AD. Formation, Structure and Reactivity of Copper Dioxygen Complexes. In: Reedijk J, Bouwman E, editors. Bioinorganic Catalysis. 2. Marcel Dekker, Inc; New York: 1999. pp. 469–534. Revised and Expanded.
- Quant Hatcher L, Karlin KD. J Biol Inorg Chem. 2004;9:669–683. - PubMed
- Lee Y, Karlin KD. Highlights of Copper Protein Active-Site Structure/Reactivity and Synthetic Model Studies. In: Metzler-Nolte N, Kraatz H-B, editors. Concepts and Models in Bioinorganic Chemistry. Wiley-VCH; New York: 2006. pp. 363–395.
-
- Klinman JP. Chem Rev. 1996;96:2541–2561. - PubMed
- Klinman JP. J Biol Chem. 2006;281:3013–3016. - PubMed
- Prigge ST, Eipper B, Mains R, Amzel LM. Science. 2004;304:864–867. - PubMed
- Chen P, Solomon EI. Proc Nat Acad Sci. 2004;101:13105–13110. - PMC - PubMed
- Balasubramanian R, Smith SM, Rawat S, Yatsunyk LA, Stemmler TL, Rosenzweig AC. Nature. 2010;465:115–119. - PMC - PubMed
- Chan SI, Yu SSF. Accounts Chem Res. 2008;41:969–979. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
