

Analysis of Cryptococcus neoformans sexual development reveals rewiring of the pheromone-response network by a change in transcription factor identity
- PMID: 22466042
- PMCID: PMC3374309
- DOI: 10.1534/genetics.112.138958
Analysis of Cryptococcus neoformans sexual development reveals rewiring of the pheromone-response network by a change in transcription factor identity
Abstract
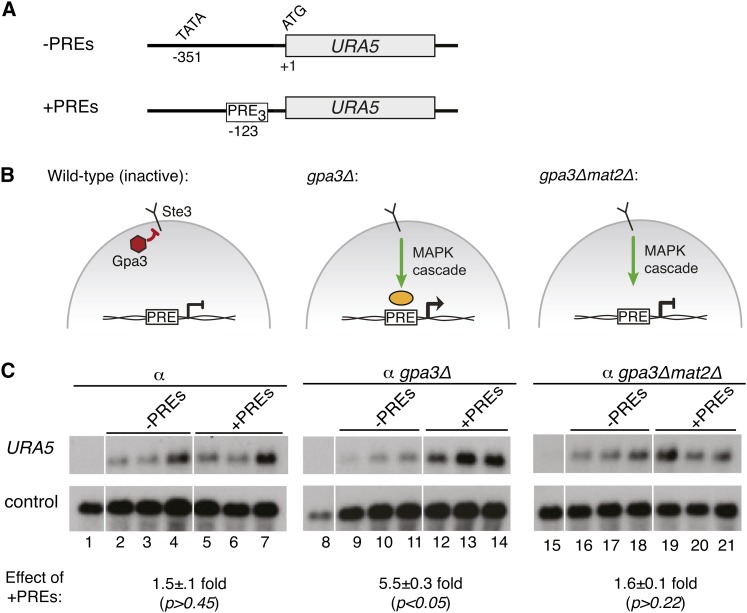
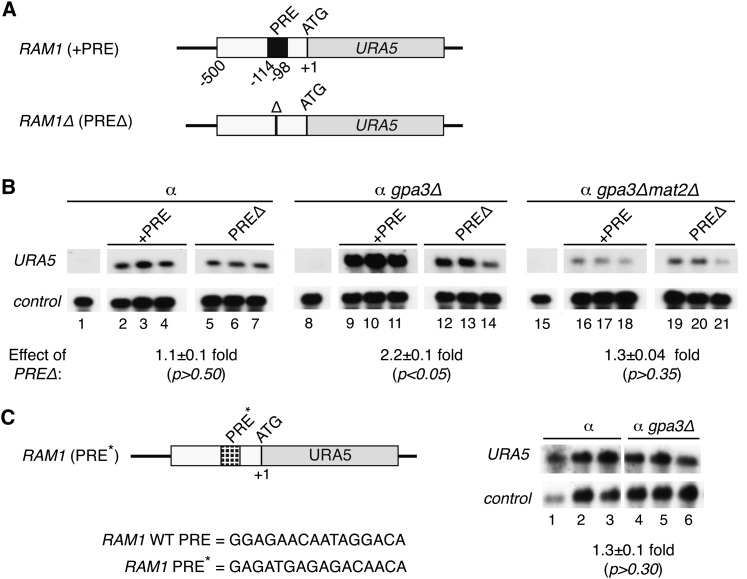
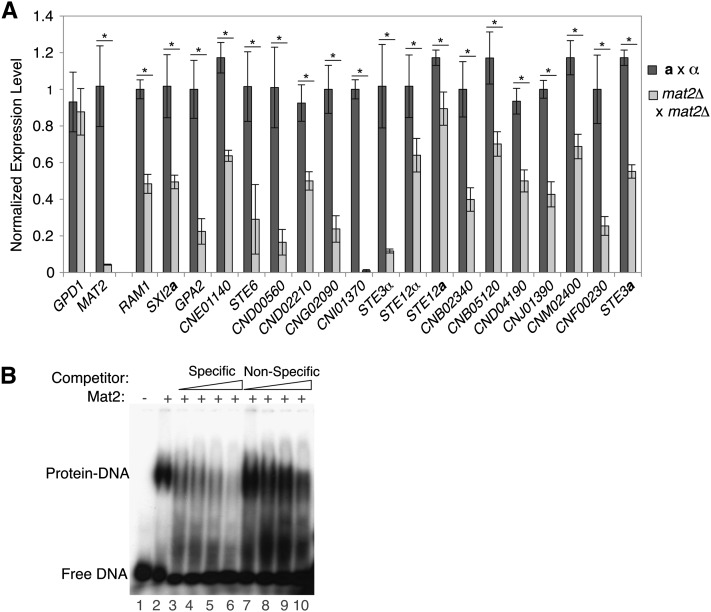
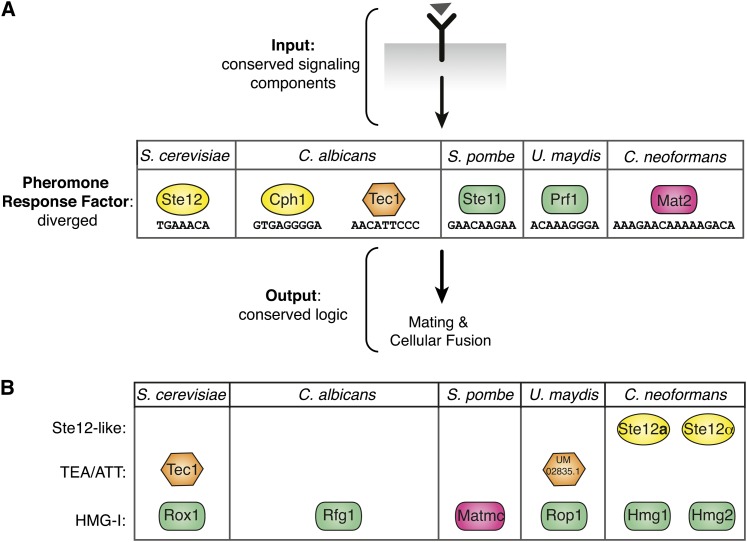
The fundamental mechanisms that control eukaryotic development include extensive regulation at the level of transcription. Gene regulatory networks, composed of transcription factors, their binding sites in DNA, and their target genes, are responsible for executing transcriptional programs. While divergence of these control networks drives species-specific gene expression that contributes to biological diversity, little is known about the mechanisms by which these networks evolve. To investigate how network evolution has occurred in fungi, we used a combination of microarray expression profiling, cis-element identification, and transcription-factor characterization during sexual development of the human fungal pathogen Cryptococcus neoformans. We first defined the major gene expression changes that occur over time throughout sexual development. Through subsequent bioinformatic and molecular genetic analyses, we identified and functionally characterized the C. neoformans pheromone-response element (PRE). We then discovered that transcriptional activation via the PRE requires direct binding of the high-mobility transcription factor Mat2, which we conclude functions as the elusive C. neoformans pheromone-response factor. This function of Mat2 distinguishes the mechanism of regulation through the PRE of C. neoformans from all other fungal systems studied to date and reveals species-specific adaptations of a fungal transcription factor that defies predictions on the basis of sequence alone. Overall, our findings reveal that pheromone-response network rewiring has occurred at the level of transcription factor identity, despite the strong conservation of upstream and downstream components, and serve as a model for how selection pressures act differently on signaling vs. gene regulatory components during eukaryotic evolution.
Figures
References
-
- Alspaugh J., Davidson R., Heitman J., 2000. Morphogenesis of Cryptococcus neoformans. Contrib. Microbiol. 5: 217–238 - PubMed
-
- Ausubel F., Brent R., Kingston R., Moore D., Seidman J., et al. (Editors), 1997. Current Protocols in Molecular Biology. Wiley, New York
-
- Bailey T. L., Elkan C., 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers, 28–36 Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. AAAI Press, Palo Alto, CA - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
