

NKG2D mediates NK cell hyperresponsiveness and influenza-induced pathologies in a mouse model of chronic obstructive pulmonary disease
- PMID: 22467655
- PMCID: PMC3331972
- DOI: 10.4049/jimmunol.1102643
NKG2D mediates NK cell hyperresponsiveness and influenza-induced pathologies in a mouse model of chronic obstructive pulmonary disease
Abstract
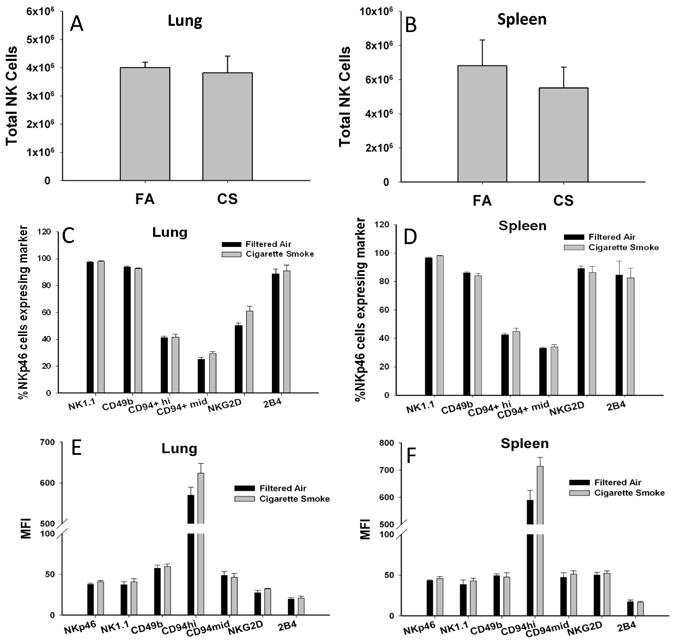
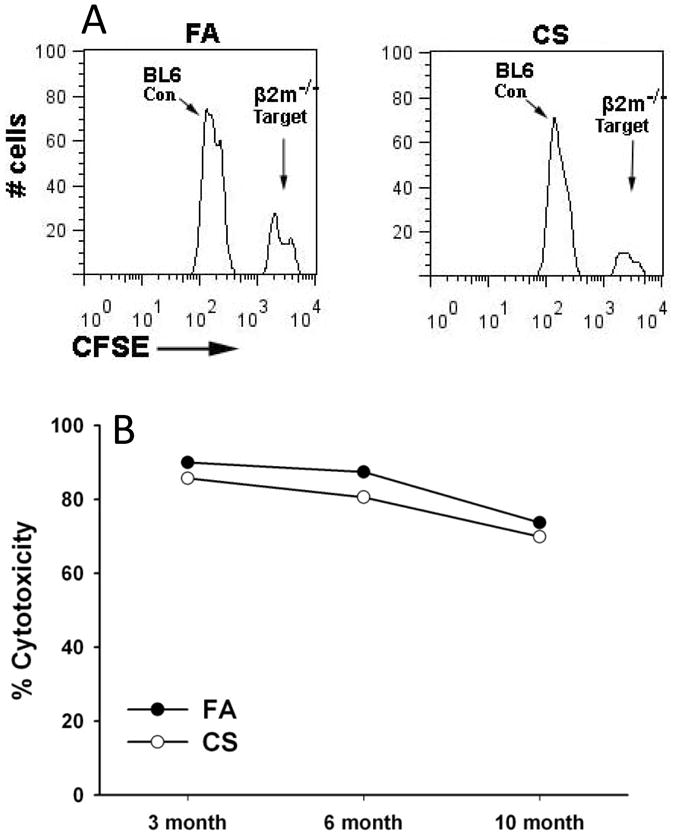
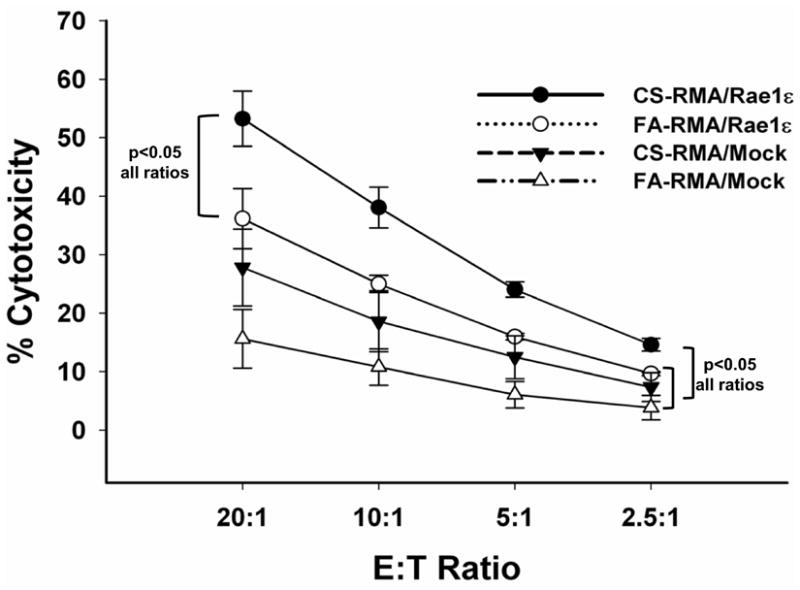
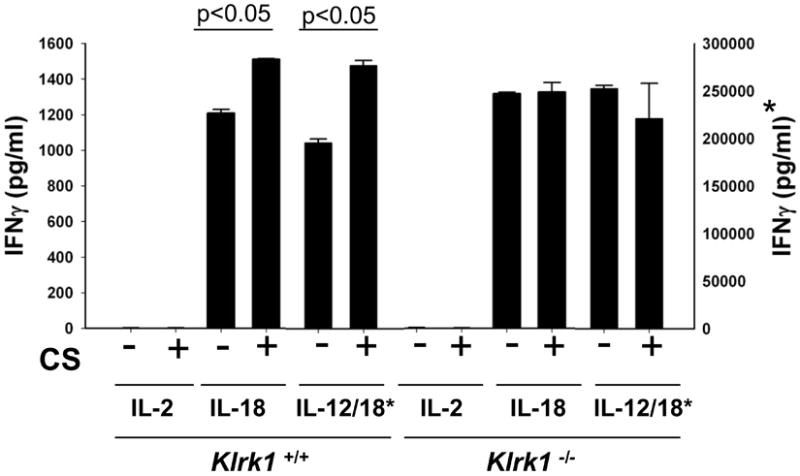
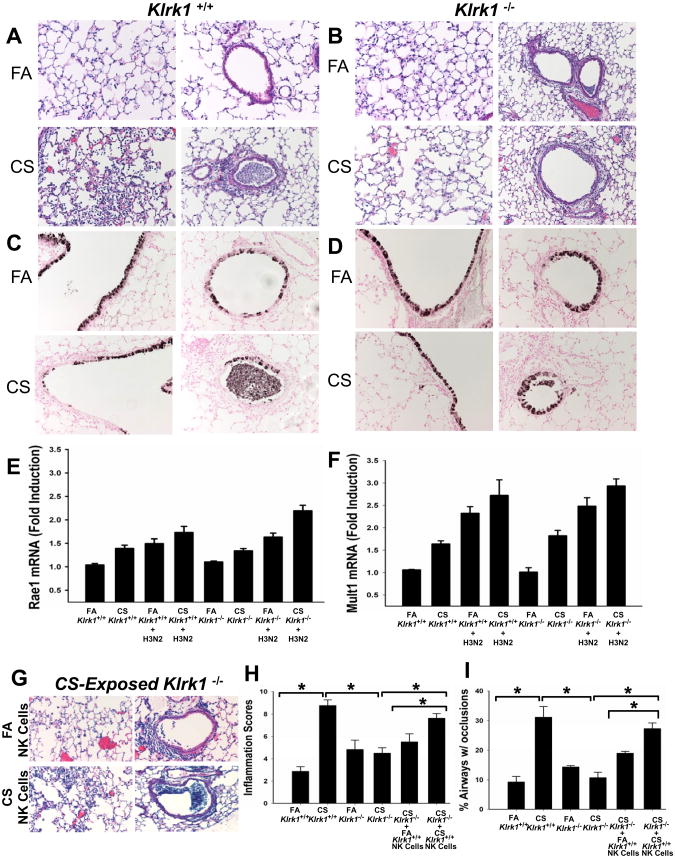
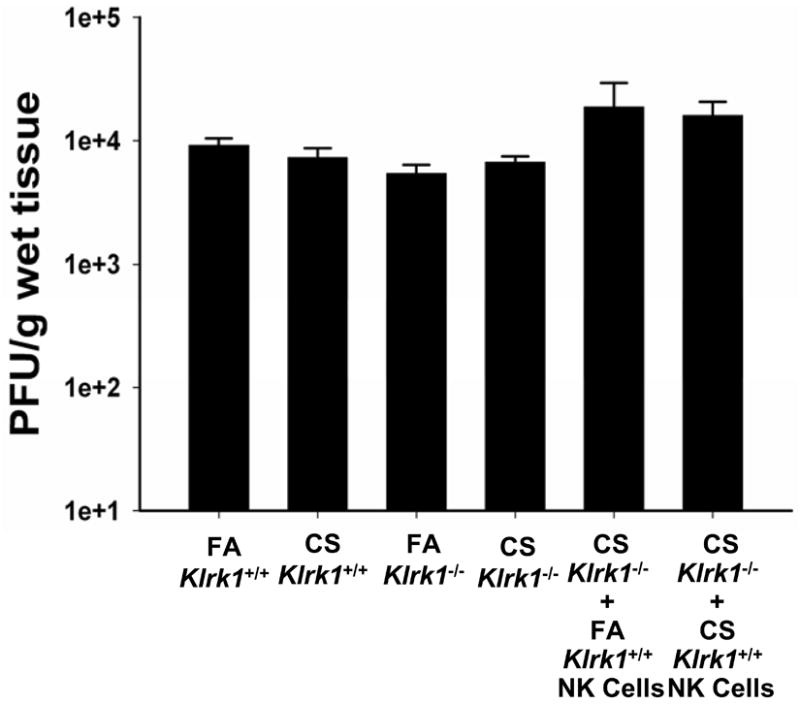
Chronic obstructive pulmonary disease (COPD) is characterized by peribronchial and perivascular inflammation and largely irreversible airflow obstruction. Acute disease exacerbations, due frequently to viral infections, lead to enhanced disease symptoms and contribute to long-term progression of COPD pathology. Previously, we demonstrated that NK cells from cigarette smoke (CS)-exposed mice exhibit enhanced effector functions in response to stimulating cytokines or TLR ligands. In this article, we show that the activating receptor NKG2D is a key mediator for CS-stimulated NK cell hyperresponsiveness, because CS-exposed NKG2D-deficient mice (Klrk1(-/-)) did not exhibit enhanced effector functions as assessed by cytokine responsiveness. NK cell cytotoxicity against MHC class I-deficient targets was not affected in a COPD model. However, NK cells from CS-exposed mice exhibit greater cytotoxic activity toward cells that express the NKG2D ligand RAET1ε. We also demonstrate that NKG2D-deficient mice exhibit diminished airway damage and reduced inflammation in a model of viral COPD exacerbation, which do not affect viral clearance. Furthermore, adoptive transfer of NKG2D(+) NK cells into CS-exposed, influenza-infected NKG2D-deficient mice recapitulated the phenotypes observed in CS-exposed, influenza-infected wild-type mice. Our findings indicate that NKG2D stimulation during long-term CS exposure is a central pathway in the development of NK cell hyperresponsiveness and influenza-mediated exacerbations of COPD.
Figures
Similar articles
-
TLR and NKG2D signaling pathways mediate CS-induced pulmonary pathologies.PLoS One. 2013 Oct 9;8(10):e78735. doi: 10.1371/journal.pone.0078735. eCollection 2013. PLoS One. 2013. PMID: 24130907 Free PMC article.
-
Chronic cigarette smoke exposure primes NK cell activation in a mouse model of chronic obstructive pulmonary disease.J Immunol. 2010 Apr 15;184(8):4460-9. doi: 10.4049/jimmunol.0903654. Epub 2010 Mar 12. J Immunol. 2010. PMID: 20228194
-
Critical role of the NKG2D receptor for NK cell-mediated control and immune escape of B-cell lymphoma.Eur J Immunol. 2015 Sep;45(9):2593-601. doi: 10.1002/eji.201445375. Epub 2015 Jul 7. Eur J Immunol. 2015. PMID: 26151313
-
Role of Polymorphisms of NKG2D Receptor and Its Ligands in Acute Myeloid Leukemia and Human Stem Cell Transplantation.Front Immunol. 2021 Mar 30;12:651751. doi: 10.3389/fimmu.2021.651751. eCollection 2021. Front Immunol. 2021. PMID: 33868289 Free PMC article. Review.
-
Murine NKG2D ligands: "double, double toil and trouble".Mol Immunol. 2009 Mar;46(6):1011-9. doi: 10.1016/j.molimm.2008.09.035. Epub 2008 Dec 10. Mol Immunol. 2009. PMID: 19081632 Free PMC article. Review.
Cited by
-
Role of ribonuclease L in viral pathogen-associated molecular pattern/influenza virus and cigarette smoke-induced inflammation and remodeling.J Immunol. 2013 Sep 1;191(5):2637-46. doi: 10.4049/jimmunol.1300082. Epub 2013 Aug 2. J Immunol. 2013. PMID: 23913960 Free PMC article.
-
Role of CC chemokine receptor 4 in natural killer cell activation during acute cigarette smoke exposure.Am J Pathol. 2014 Feb;184(2):454-63. doi: 10.1016/j.ajpath.2013.10.017. Epub 2013 Dec 9. Am J Pathol. 2014. PMID: 24333113 Free PMC article.
-
1H NMR-based profiling reveals differential immune-metabolic networks during influenza virus infection in obese mice.PLoS One. 2014 May 20;9(5):e97238. doi: 10.1371/journal.pone.0097238. eCollection 2014. PLoS One. 2014. PMID: 24844920 Free PMC article.
-
Human CD56+ cytotoxic lung lymphocytes kill autologous lung cells in chronic obstructive pulmonary disease.PLoS One. 2014 Jul 31;9(7):e103840. doi: 10.1371/journal.pone.0103840. eCollection 2014. PLoS One. 2014. PMID: 25078269 Free PMC article.
-
The Paradoxical Role of NKG2D in Cancer Immunity.Front Immunol. 2018 Aug 13;9:1808. doi: 10.3389/fimmu.2018.01808. eCollection 2018. Front Immunol. 2018. PMID: 30150983 Free PMC article. Review.
References
-
- Pauwels R. Global initiative for chronic obstructive lung diseases (GOLD): time to act. Eur Respir J. 2001;18:901–902. - PubMed
-
- Rennard SI, Vestbo J. COPD: the dangerous underestimate of 15% Lancet. 2006;367:1216–1219. - PubMed
-
- Rodriguez-Roisin R. Toward a consensus definition for COPD exacerbations. Chest. 2000;117:398S–401S. - PubMed
-
- Wedzicha JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2004;1:115–120. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
