

Solution structural ensembles of substrate-free cytochrome P450(cam)
- PMID: 22468842
- PMCID: PMC3424371
- DOI: 10.1021/bi300007r
Solution structural ensembles of substrate-free cytochrome P450(cam)
Abstract
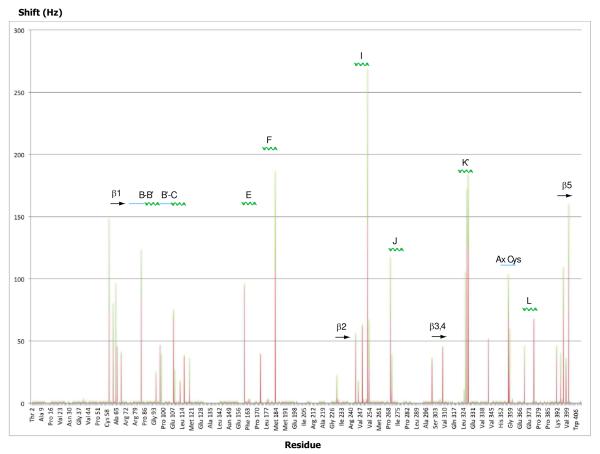
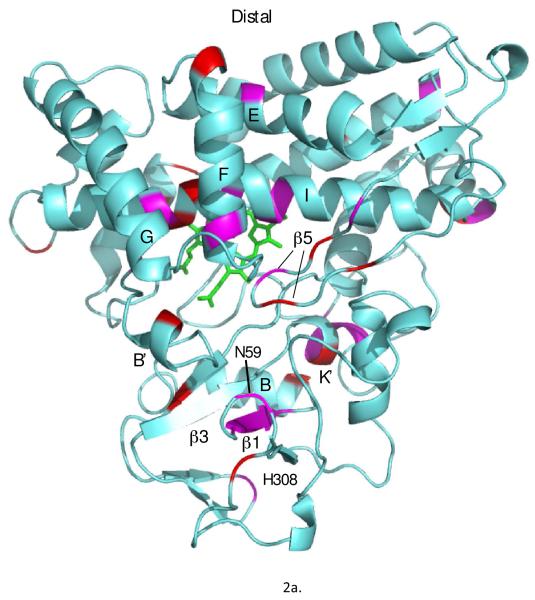
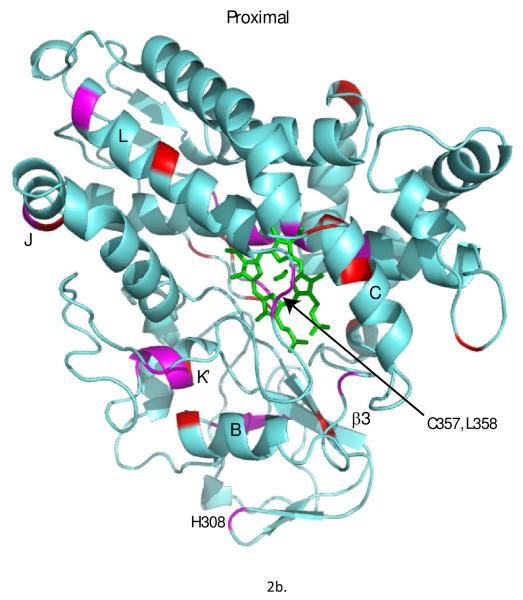
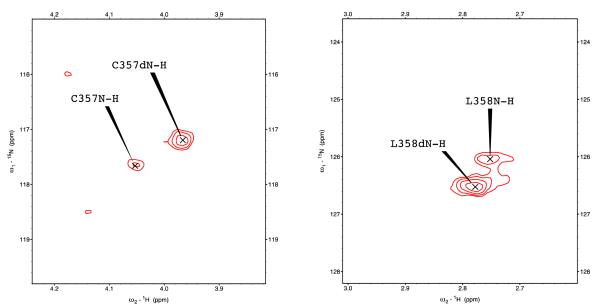
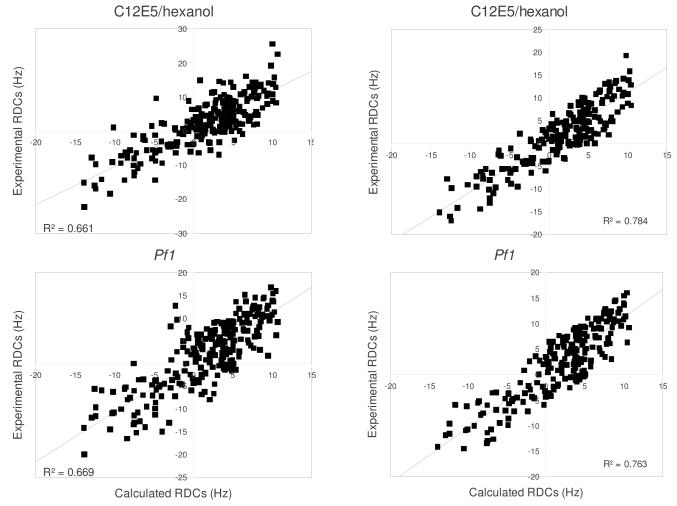
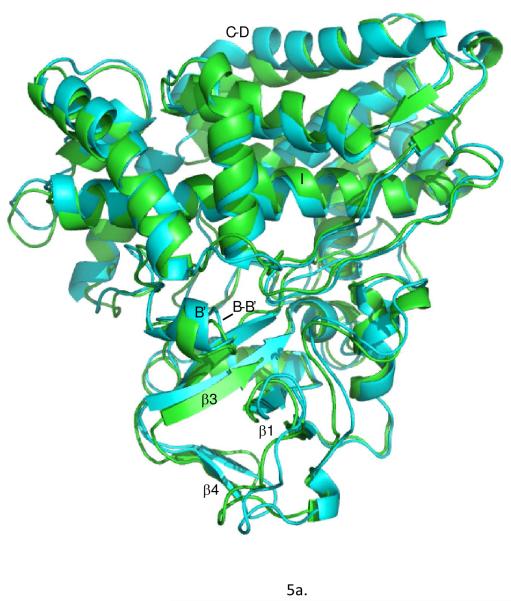
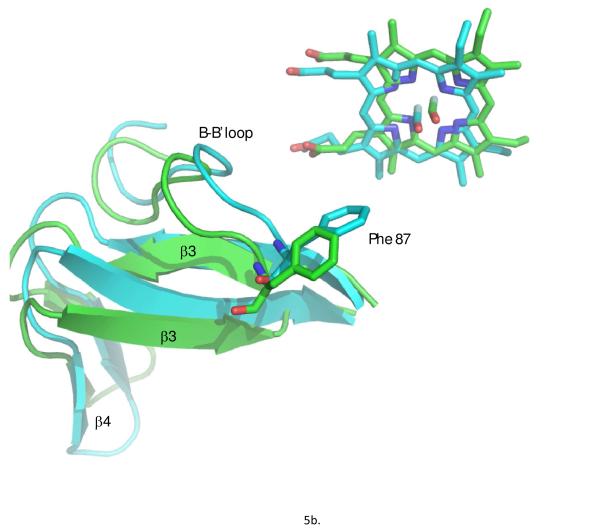
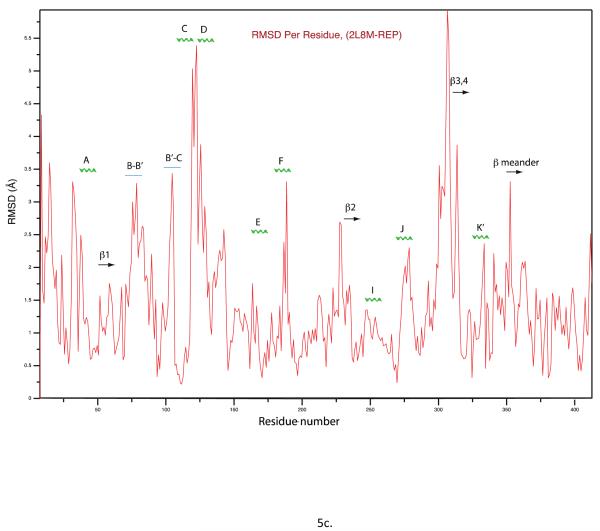
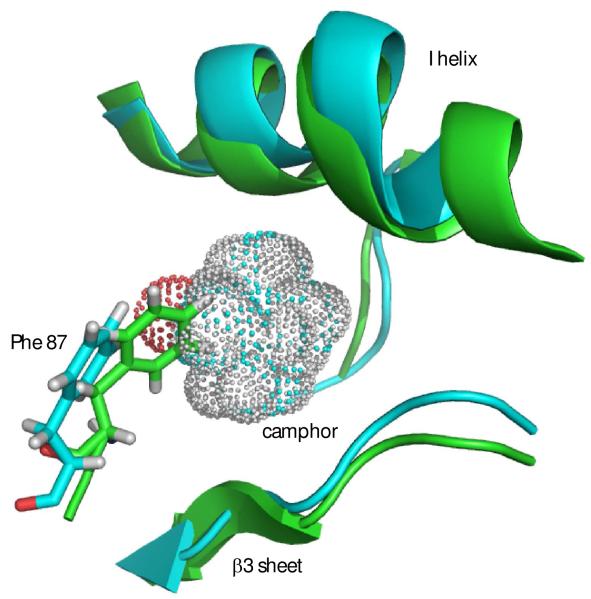
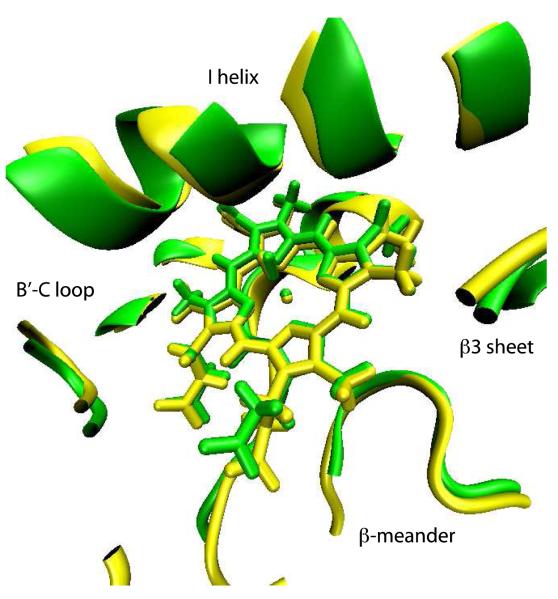
Removal of substrate (+)-camphor from the active site of cytochrome P450(cam) (CYP101A1) results in nuclear magnetic resonance-detected perturbations in multiple regions of the enzyme. The (1)H-(15)N correlation map of substrate-free diamagnetic Fe(II) CO-bound CYP101A permits these perturbations to be mapped onto the solution structure of the enzyme. Residual dipolar couplings (RDCs) were measured for (15)N-(1)H amide pairs in two independent alignment media for the substrate-free enzyme and used as restraints in solvated molecular dynamics (MD) simulations to generate an ensemble of best-fit structures of the substrate-free enzyme in solution. Nuclear magnetic resonance-detected chemical shift perturbations reflect changes in the electronic environment of the NH pairs, such as hydrogen bonding and ring current shifts, and are observed for residues in the active site as well as in hinge regions between secondary structural features. RDCs provide information about relative orientations of secondary structures, and RDC-restrained MD simulations indicate that portions of a β-rich region adjacent to the active site shift so as to partially occupy the vacancy left by removal of the substrate. The accessible volume of the active site is reduced in the substrate-free enzyme relative to the substrate-bound structure calculated using the same methods. Both symmetric and asymmetric broadening of multiple resonances observed upon substrate removal as well as localized increased errors in RDC fits suggest that an ensemble of enzyme conformations are present in the substrate-free form.
Figures
References
-
- Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber. Deutsche Chem. Gessel. 1894;27:2985–2993.
-
- Niesen MJM, Bhattacharya S, Vaidehi N. The role of conformational ensembles in ligand recognition in G-protein coupled receptors. J. Am. Chem. Soc. 2011;133:13197–13204. - PubMed
-
- Frauenfelder H, Petsko GA, Tsernoglou D. Temperature-dependent X-ray diffraction as a probe of protein structural dynamics. Nature. 1979;280:558–563. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
